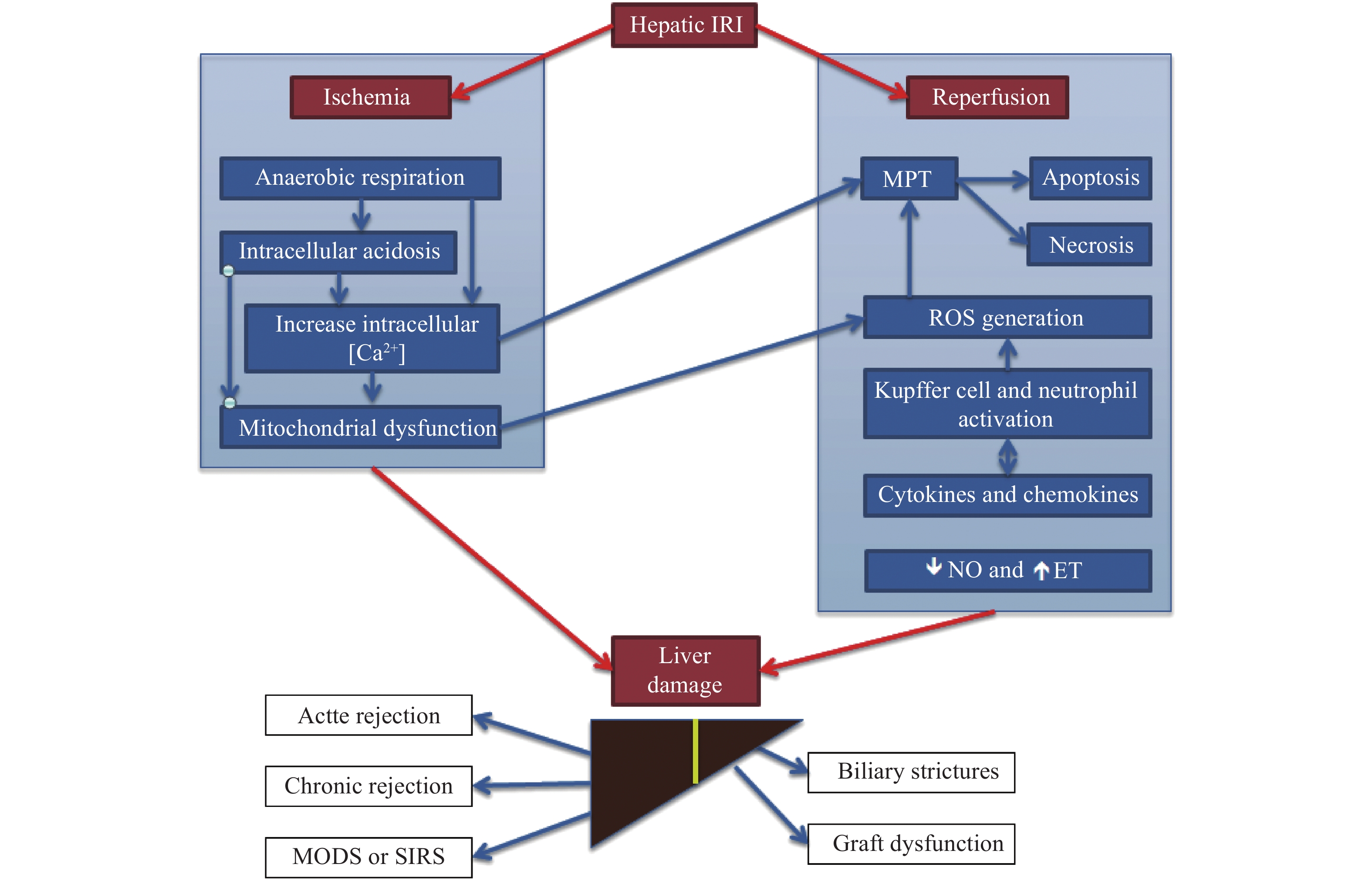
| [1] |
Ikeda T, Yanaga K, Kishikawa K, et al. Ischemic injury in liver transplantation: difference in injury sites between warm and cold ischemia in rats[J]. Hepatology, 1992, 16(2): 454–461. doi: 10.1002/hep.1840160226
|
| [2] |
Jaeschke H. Reperfusion injury after warm ischemia or cold storage of the liver: role of apoptotic cell death[J]. Transplant Proc, 2002, 34(7): 2656–2658. doi: 10.1016/S0041-1345(02)03464-4
|
| [3] |
Huet PM, Nagaoka MR, Desbiens G, et al. Sinusoidal endothelial cell and hepatocyte death following cold ischemia-warm reperfusion of the rat liver[J]. Hepatology, 2004, 39(4): 1110–1119. doi: 10.1002/hep.20157
|
| [4] |
Kupiec-Weglinski JW, Busuttil RW. Ischemia and reperfusion injury in liver transplantation[J]. Transplant Proc, 2005, 37(4): 1653–1656. doi: 10.1016/j.transproceed.2005.03.134
|
| [5] |
Zhai Y, Busuttil RW, Kupiec-Weglinski JW. Liver ischemia and reperfusion injury: new insights into mechanisms of innate-adaptive immune-mediated tissue inflammation[J]. Am J Transplant, 2011, 11(8): 1563–1569. doi: 10.1111/ajt.2011.11.issue-8
|
| [6] |
Pine JK, Aldouri A, Young AL, et al. Liver transplantation following donation after cardiac death: an analysis using matched pairs[J]. Liver Transpl, 2009, 15(9): 1072–1082. doi: 10.1002/lt.v15:9
|
| [7] |
Howard TK, Klintmalm GBG, Cofer JB, et al. The influence of preservation injury on rejection in the hepatic transplant recipient[J]. Transplantation, 1990, 49(1): 103–107. doi: 10.1097/00007890-199001000-00023
|
| [8] |
Fellström B, Aküyrek LM, Backman U, et al. Postischemic reperfusion injury and allograft arteriosclerosis[J]. Transplant Proc, 1998, 30(8): 4278–4280. doi: 10.1016/S0041-1345(98)01412-2
|
| [9] |
Guo WA. The search for a magic bullet to fight multiple organ failure secondary to ischemia/reperfusion injury and abdominal compartment syndrome[J]. J Surg Res, 2013, 184(2): 792–793. doi: 10.1016/j.jss.2012.06.024
|
| [10] |
Wertheim JA, Petrowsky H, Saab S, et al. Major challenges limiting liver transplantation in the United States[J]. Am J Transplant, 2011, 11(9): 1773–1784. doi: 10.1111/j.1600-6143.2011.03587.x
|
| [11] |
Neuberger J. Liver transplantation in the United Kingdom[J]. Liver Transpl, 2016, 22(8): 1129–1135. doi: 10.1002/lt.v22.8
|
| [12] |
NHS Blood and Transplant. Annual activity report[EB/OL]. [2017-02-07]. www.odt.nhs.uk.
|
| [13] |
Singal AK, Guturu P, Hmoud B, et al. Evolving frequency and outcomes of liver transplantation based on etiology of liver disease[J]. Transplantation, 2013, 95(5): 755–760. doi: 10.1097/TP.0b013e31827afb3a
|
| [14] |
Casillas-Ramírez A, Mosbah IB, Ramalho F, et al. Past and future approaches to ischemia-reperfusion lesion associated with liver transplantation[J]. Life Sci, 2006, 79(20): 1881–1894. doi: 10.1016/j.lfs.2006.06.024
|
| [15] |
Fan CG, Zwacka RM, Engelhardt JF. Therapeutic approaches for ischemia/reperfusion injury in the liver[J]. J Mol Med (Berl), 1999, 77(8): 577–592. doi: 10.1007/s001099900029
|
| [16] |
Zwacka RM, Zhou WH, Zhang YL, et al. Redox gene therapy for ischemia/reperfusion injury of the liver reduces AP1 and NF-κB activation[J]. Nat Med, 1998, 4(6): 698–704. doi: 10.1038/nm0698-698
|
| [17] |
Teoh NC, Farrell GC. Hepatic ischemia reperfusion injury: pathogenic mechanisms and basis for hepatoprotection[J]. J Gastroenterol Hepatol, 2003, 18(8): 891–902. doi: 10.1046/j.1440-1746.2003.03056.x
|
| [18] |
Mavier P, Preaux AM, Guigui B, et al. In vitro toxicity of polymorphonuclear neutrophils to rat hepatocytes: evidence for a proteinase-mediated mechanism[J]. Hepatology, 1988, 8(2): 254–258. doi: 10.1002/hep.1840080211
|
| [19] |
Li XK, Matin AF, Suzuki H, et al. Effect of protease inhibitor on ischemia/reperfusion injury of the rat liver[J]. Transplantation, 1993, 56(6): 1331–1336. doi: 10.1097/00007890-199312000-00008
|
| [20] |
Nastos C, Kalimeris K, Papoutsidakis N, et al. Global consequences of liver ischemia/reperfusion injury[J]. Oxid Med Cell Longev, 2014, 2014: 906965.
|
| [21] |
Selzner M, Selzner N, Jochum W, et al. Increased ischemic injury in old mouse liver: an ATP-dependent mechanism[J]. Liver Transpl, 2007, 13(3): 382–390. doi: 10.1002/lt.21100
|
| [22] |
Wang D, Dou K, Song Z, et al. The Na(+)/H(+) exchange inhibitor: a new therapeutic approach for hepatic ischemia injury in rats[J]. Transplant Proc, 2003, 35(8): 3134–3135. doi: 10.1016/j.transproceed.2003.10.021
|
| [23] |
Carini R, De Cesaris MG, Splendore R, et al. Alterations of Na+ homeostasis in hepatocyte reoxygenation injury[J]. Biochim Biophys Acta, 2000, 1500(3): 297–305. doi: 10.1016/S0925-4439(99)00114-3
|
| [24] |
Nishimura Y, Romer LH, Lemasters JJ. Mitochondrial dysfunction and cytoskeletal disruption during chemical hypoxia to cultured rat hepatic sinusoidal endothelial cells: the pH paradox and cytoprotection by glucose, acidotic pH, and glycine[J]. Hepatology, 1998, 27(4): 1039–1049. doi: 10.1002/hep.510270420
|
| [25] |
Vairetti M, Richelmi P, Bertè F, et al. Role of pH in protection by low sodium against hypoxic injury in isolated perfused rat livers[J]. J Hepatol, 2006, 44(5): 894–901. doi: 10.1016/j.jhep.2005.08.007
|
| [26] |
Gores GJ, Nieminen AL, Wray BE, et al. Intracellular pH during " chemical hypoxia” in cultured rat hepatocytes. Protection by intracellular acidosis against the onset of cell death[J]. J Clin Invest, 1989, 83(2): 386–396. doi: 10.1172/JCI113896
|
| [27] |
Jiang N, Zhang ZM, Liu L, et al. Effects of Ca 2+ channel blockers on store-operated Ca 2+ channel currents of Kupffer cells after hepatic ischemia/reperfusion injury in rats[J]. World J Gastroenterol, 2006, 12(29): 4694–4698. doi: 10.3748/wjg.v12.i29.4694
|
| [28] |
Barritt GJ, Chen JL, Rychkov GY. Ca 2+-permeable channels in the hepatocyte plasma membrane and their roles in hepatocyte physiology[J]. Biochim Biophys Acta, 2008, 1783(5): 651–672. doi: 10.1016/j.bbamcr.2008.01.016
|
| [29] |
Wang HG, Pathan N, Ethell IM, et al. Ca 2+-induced apoptosis through calcineurin dephosphorylation of BAD[J]. Science, 1999, 284(5412): 339–343. doi: 10.1126/science.284.5412.339
|
| [30] |
Anderson CD, Pierce J, Nicoud I, et al. Modulation of mitochondrial calcium management attenuates hepatic warm ischemia-reperfusion injury[J]. Liver Transpl, 2005, 11(6): 663–668. doi: 10.1002/lt.20407
|
| [31] |
Jaeschke H, Lemasters JJ. Apoptosis versus oncotic necrosis in hepatic ischemia/reperfusion injury[J]. Gastroenterology, 2003, 125(4): 1246–1257. doi: 10.1016/S0016-5085(03)01209-5
|
| [32] |
Nauta RJ, Tsimoyiannis E, Uribe M, et al. The role of calcium ions and calcium channel entry blockers in experimental ischemia-reperfusion-induced liver injury[J]. Ann Surg, 1991, 213(2): 137–142. doi: 10.1097/00000658-199102000-00008
|
| [33] |
Hataji K, Watanabe T, Oowada S, et al. Effects of a calcium-channel blocker (CV159) on hepatic ischemia/reperfusion injury in rats: evaluation with selective NO/pO2 electrodes and an electron paramagnetic resonance spin-trapping method[J]. Biol Pharm Bull, 2010, 33(1): 77–83. doi: 10.1248/bpb.33.77
|
| [34] |
Nicoud IB, Knox CD, Jones CM, et al. 2-APB protects against liver ischemia-reperfusion injury by reducing cellular and mitochondrial calcium uptake[J]. Am J Physiol Gastrointest Liver Physiol, 2007, 293(3): G623–G630. doi: 10.1152/ajpgi.00521.2006
|
| [35] |
Pronobesh C, Dagagi AV, Pallab C, et al. Protective role of the calcium channel blocker amlodipine against mitochondrial injury in ischemia and reperfusion injury of rat liver[J]. Acta Pharm, 2008, 58(4): 421–428.
|
| [36] |
Abu-Amara M, Yang SY, Tapuria N, et al. Liver ischemia/reperfusion injury: processes in inflammatory networks—a review[J]. Liver Transpl, 2010, 16(9): 1016–1032. doi: 10.1002/lt.22117
|
| [37] |
Elmore SP, Qian T, Grissom SF, et al. The mitochondrial permeability transition initiates autophagy in rat hepatocytes[J]. FASEB J, 2001, 15(12): 2286–2287. doi: 10.1096/fj.01-0206fje
|
| [38] |
Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy[J]. Arch Biochem Biophys, 2007, 462(2): 245–253. doi: 10.1016/j.abb.2007.03.034
|
| [39] |
Zhao KS, Zhao GM, Wu DL, et al. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury[J]. J Biol Chem, 2004, 279(33): 34682–34690. doi: 10.1074/jbc.M402999200
|
| [40] |
Kim JS, Qian T, Lemasters JJ. Mitochondrial permeability transition in the switch from necrotic to apoptotic cell death in ischemic rat hepatocytes[J]. Gastroenterology, 2003, 124(2): 494–503. doi: 10.1053/gast.2003.50059
|
| [41] |
Sastre J, Serviddio G, Pereda J, et al. Mitochondrial function in liver disease[J]. Front Biosci, 2007, 12: 1200–1209. doi: 10.2741/2138
|
| [42] |
Videla LA. Cytoprotective and suicidal signaling in oxidative stress[J]. Biol Res, 2010, 43(3): 363–369.
|
| [43] |
Hines IN, Hoffman JM, Scheerens H, et al. Regulation of postischemic liver injury following different durations of ischemia[J]. Am J Physiol Gastrointest Liver Physiol, 2003, 284(3): G536–G545. doi: 10.1152/ajpgi.00400.2002
|
| [44] |
Jaeschke H. Mechanisms of Liver Injury. II. Mechanisms of neutrophil-induced liver cell injury during hepatic ischemia-reperfusion and other acute inflammatory conditions[J]. Am J Physiol Gastrointest Liver Physiol, 2006, 290(6): G1083–G1088. doi: 10.1152/ajpgi.00568.2005
|
| [45] |
Spencer NY, Zhou WH, Li Q, et al. Hepatocytes produce TNF-α following hypoxia-reoxygenation and liver ischemia-reperfusion in a NADPH oxidase- and c-Src-dependent manner[J]. Am J Physiol Gastrointest Liver Physiol, 2013, 305(1): G84–G94. doi: 10.1152/ajpgi.00430.2012
|
| [46] |
Reiniers MJ, van Golen RF, van Gulik TM, et al. Reactive oxygen and nitrogen species in steatotic hepatocytes: a molecular perspective on the pathophysiology of ischemia-reperfusion injury in the fatty liver[J]. Antioxid Redox Signal, 2014, 21(7): 1119–1142. doi: 10.1089/ars.2013.5486
|
| [47] |
Pardini RS. Toxicity of oxygen from naturally occurring redox-active pro-oxidants[J]. Arch Insect Biochem Physiol, 1995, 29(2): 101–118. doi: 10.1002/arch.940290203
|
| [48] |
Jaeschke H. Reactive oxygen and mechanisms of inflammatory liver injury: present concepts[J]. J Gastroenterol Hepatol, 2011, 26(S1): 173–179.
|
| [49] |
Guicciardi ME, Malhi H, Mott JL, et al. Apoptosis and necrosis in the liver[J]. Compr Physiol, 2013, 3(2): 977–1010.
|
| [50] |
Rauen U, Polzar B, Stephan H, et al. Cold-induced apoptosis in cultured hepatocytes and liver endothelial cells: mediation by reactive oxygen species[J]. FASEB J, 1999, 13(1): 155–168. doi: 10.1096/fasebj.13.1.155
|
| [51] |
Kawada N, Tran-Thi TA, Klein H, et al. The contraction of hepatic stellate (Ito) cells stimulated with vasoactive substances: Possible involvement of endothelin 1 and nitric oxide in the regulation of the sinusoidal tonus[J]. Eur J Biochem, 1993, 213(2): 815–823. doi: 10.1111/ejb.1993.213.issue-2
|
| [52] |
Kawamura E, Yamanaka N, Okamoto E, et al. Response of plasma and tissue endothelin-1 to liver ischemia and its implication in ischemia-reperfusion injury[J]. Hepatology, 1995, 21(4): 1138–1143. doi: 10.1016/0270-9139(95)90266-X
|
| [53] |
Lefer AM, Lefer DJ. Nitric oxide. II. Nitric oxide protects in intestinal inflammation[J]. Am J Physiol, 1999, 276(3 Pt 1): G572–G575.
|
| [54] |
Hamada T, Duarte S, Tsuchihashi S, et al. Inducible nitric oxide synthase deficiency impairs matrix metalloproteinase-9 activity and disrupts leukocyte migration in hepatic ischemia/reperfusion injury[J]. Am J Pathol, 2009, 174(6): 2265–2277. doi: 10.2353/ajpath.2009.080872
|
| [55] |
Abu-Amara M, Yang SY, Seifalian A, et al. The nitric oxide pathway-evidence and mechanisms for protection against liver ischaemia reperfusion injury[J]. Liver Int, 2012, 32(4): 531–543. doi: 10.1111/liv.2012.32.issue-4
|
| [56] |
Chen C, Lee WH, Zhong LW, et al. Regulatory T cells can mediate their function through the stimulation of APCs to produce immunosuppressive nitric oxide[J]. J Immunol, 2006, 176(6): 3449–3460. doi: 10.4049/jimmunol.176.6.3449
|
| [57] |
Phillips L, Toledo AH, Lopez-Neblina F, et al. Nitric oxide mechanism of protection in ischemia and reperfusion injury[J]. J Invest Surg, 2009, 22(1): 46–55. doi: 10.1080/08941930802709470
|
| [58] |
Lang JD Jr, Teng XJ, Chumley P, et al. Inhaled NO accelerates restoration of liver function in adults following orthotopic liver transplantation[J]. J Clin Invest, 2007, 117(9): 2583–2591. doi: 10.1172/JCI31892
|
| [59] |
Duranski MR, Greer JJM, Dejam A, et al. Cytoprotective effects of nitrite during in vivo ischemia-reperfusion of the heart and liver[J]. J Clin Invest, 2005, 115(5): 1232–1240. doi: 10.1172/JCI22493
|
| [60] |
Li W, Meng ZH, Liu YL, et al. The hepatoprotective effect of sodium nitrite on cold ischemia-reperfusion injury[J]. J Transplant, 2012, 2012: 635179.
|
| [61] |
Shiratori Y, Kiriyama H, Fukushi Y, et al. Modulation of ischemia-reperfusion-induced hepatic injury by Kupffer cells[J]. Dig Dis Sci, 1994, 39(6): 1265–1272. doi: 10.1007/BF02093792
|
| [62] |
Jaeschke H, Bautista AP, Spolarics Z, et al. Superoxide generation by neutrophils and Kupffer cells during in vivo reperfusion after hepatic ischemia in rats[J]. J Leukoc Biol, 1992, 52(4): 377–382. doi: 10.1002/jlb.1992.52.issue-4
|
| [63] |
Fondevila C, Shen XD, Tsuchihashi S, et al. The membrane attack complex (C5b-9) in liver cold ischemia and reperfusion injury[J]. Liver Transpl, 2008, 14(8): 1133–1141. doi: 10.1002/lt.v14:8
|
| [64] |
Brock RW, Nie RG, Harris KA, et al. Kupffer cell-initiated remote hepatic injury following bilateral hindlimb ischemia is complement dependent[J]. Am J Physiol Gastrointest Liver Physiol, 2001, 280(2): G279–G284. doi: 10.1152/ajpgi.2001.280.2.G279
|
| [65] |
Llacuna L, Marí M, Lluis JM, et al. Reactive oxygen species mediate liver injury through parenchymal nuclear factor-κB inactivation in prolonged ischemia/reperfusion[J]. Am J Pathol, 2009, 174(5): 1776–1785. doi: 10.2353/ajpath.2009.080857
|
| [66] |
Selzner N, Selzner M, Odermatt B, et al. ICAM-1 triggers liver regeneration through leukocyte recruitment and Kupffer cell-dependent release of TNF-α/IL-6 in mice[J]. Gastroenterology, 2003, 124(3): 692–700. doi: 10.1053/gast.2003.50098
|
| [67] |
Boury NM, Czuprynski CJ. Listeria monocytogenes infection increases neutrophil adhesion and damage to a murine hepatocyte cell line in vitro[J]. Immunol Lett, 1995, 46(1–2): 111–116. doi: 10.1016/0165-2478(95)00027-3
|
| [68] |
Hanschen M, Zahler S, Krombach F, et al. Reciprocal activation between CD4+ T cells and Kupffer cells during hepatic ischemia-reperfusion[J]. Transplantation, 2008, 86(5): 710–718. doi: 10.1097/TP.0b013e3181821aa7
|
| [69] |
Nishimura Y, Takei Y, Kawano S, et al. The F(ab’)2 fragment of an anti-ICAM-1 monoclonal antibody attenuates liver injury after orthotopic liver transplantation[J]. Transplantation, 1996, 61(1): 99–104. doi: 10.1097/00007890-199601150-00020
|
| [70] |
Fong Y, Moldawer LL, Shires GT, et al. The biologic characteristics of cytokines and their implication in surgical injury[J]. Surg Gynecol Obstet, 1990, 170(4): 363–378.
|
| [71] |
Leifeld L, Cheng S, Ramakers J, et al. Imbalanced intrahepatic expression of interleukin 12, interferon gamma, and interleukin 10 in fulminant hepatitis B[J]. Hepatology, 2002, 36(4 Pt 1): 1001–1008.
|
| [72] |
Lentsch AB, Yoshidome H, Kato A, et al. Requirement for interleukin-12 in the pathogenesis of warm hepatic ischemia/reperfusion injury in mice[J]. Hepatology, 1999, 30(6): 1448–1453. doi: 10.1002/hep.510300615
|
| [73] |
Husted TL, Blanchard J, Schuster R, et al. Potential role for IL-23 in hepatic ischemia/reperfusion injury[J]. Inflamm Res, 2006, 55(5): 177–178. doi: 10.1007/s00011-006-0073-1
|
| [74] |
Colletti LM, Remick DG, Burtch GD, et al. Role of tumor necrosis factor-alpha in the pathophysiologic alterations after hepatic ischemia/reperfusion injury in the rat[J]. J Clin Invest, 1990, 85(6): 1936–1943. doi: 10.1172/JCI114656
|
| [75] |
Colletti LM, Kunkel SL, Walz A, et al. Chemokine expression during hepatic ischemia/reperfusion-induced lung injury in the rat. The role of epithelial neutrophil activating protein[J]. J Clin Invest, 1995, 95(1): 134–141. doi: 10.1172/JCI117630
|
| [76] |
Colletti LM, Cortis A, Lukacs N, et al. Tumor necrosis factor up-regulates intercellular adhesion molecule 1, which is important in the neutrophil-dependent lung and liver injury associated with hepatic ischemia and reperfusion in the rat[J]. Shock, 1998, 10(3): 182–191. doi: 10.1097/00024382-199809000-00006
|
| [77] |
Shito M, Wakabayashi G, Ueda M, et al. Interleukin 1 receptor blockade reduces tumor necrosis factor production, tissue injury, and mortality after hepatic ischemia-reperfusion in the rat[J]. Transplantation, 1997, 63(1): 143–148. doi: 10.1097/00007890-199701150-00026
|
| [78] |
Djeu JY, Matsushima K, Oppenheim JJ, et al. Functional activation of human neutrophils by recombinant monocyte-derived neutrophil chemotactic factor/IL-8[J]. J Immunol, 1990, 144(6): 2205–2210.
|
| [79] |
Lentsch AB, Yoshidome H, Cheadle WG, et al. Chemokine involvement in hepatic ischemia/reperfusion injury in mice: roles for macrophage inflammatory protein-2 and Kupffer cells[J]. Hepatology, 1998, 27(2): 507–512. doi: 10.1002/hep.510270226
|
| [80] |
Ke BB, Shen XD, Lassman CR, et al. Cytoprotective and antiapoptotic effects of IL-13 in hepatic cold ischemia/reperfusion injury are heme oxygenase-1 dependent[J]. Am J Transplant, 2003, 3(9): 1076–1082. doi: 10.1034/j.1600-6143.2003.00147.x
|
| [81] |
Reiter RJ, Paredes SD, Manchester LC, et al. Reducing oxidative/nitrosative stress: a newly-discovered genre for melatonin[J]. Crit Rev Biochem Mol Biol, 2009, 44(4): 175–200. doi: 10.1080/10409230903044914
|
| [82] |
López-Burillo S, Tan DX, Rodriguez-Gallego V, et al. Melatonin and its derivatives cyclic 3-hydroxymelatonin, N1-acetyl-N2-formyl-5-methoxykynuramine and 6-methoxymelatonin reduce oxidative DNA damage induced by Fenton reagents[J]. J Pineal Res, 2003, 34(3): 178–184. doi: 10.1111/jpi.2003.34.issue-3
|
| [83] |
Barlow-Walden LR, Reiter RJ, Abe M, et al. Melatonin stimulates brain glutathione peroxidase activity[J]. Neurochem Int, 1995, 26(5): 497–502. doi: 10.1016/0197-0186(94)00154-M
|
| [84] |
Reiter RJ, Tan DX, Osuna C, et al. Actions of melatonin in the reduction of oxidative stress: a review[J]. J Biomed Sci, 2000, 7(6): 444–458. doi: 10.1007/BF02253360
|
| [85] |
Okatani Y, Wakatsuki A, Reiter RJ, et al. Protective effect of melatonin against mitochondrial injury induced by ischemia and reperfusion of rat liver[J]. Eur J Pharmacol, 2003, 469(1–3): 145–152. doi: 10.1016/S0014-2999(03)01643-1
|
| [86] |
Kireev R, Bitoun S, Cuesta S, et al. Melatonin treatment protects liver of Zucker rats after ischemia/reperfusion by diminishing oxidative stress and apoptosis[J]. Eur J Pharmacol, 2013, 701(1–3): 185–193. doi: 10.1016/j.ejphar.2012.11.038
|
| [87] |
Vairetti M, Ferrigno A, Bertone R, et al. Exogenous melatonin enhances bile flow and ATP levels after cold storage and reperfusion in rat liver: implications for liver transplantation[J]. J Pineal Res, 2005, 38(4): 223–230. doi: 10.1111/jpi.2005.38.issue-4
|
| [88] |
De Deken J, Rex S, Monbaliu D, et al. The efficacy of noble gases in the attenuation of ischemia reperfusion injury: a systematic review and meta-analyses[J]. Crit Care Med, 2016, 44(9): e886–e896. doi: 10.1097/CCM.0000000000001717
|
| [89] |
Wilke HJ, Moench C, Lotz G, et al. Xenon anesthesia for liver transplant surgery: a report of four cases[J]. Transplant Proc, 2011, 43(7): 2683–2686. doi: 10.1016/j.transproceed.2011.06.029
|
| [90] |
Thies JC, Teklote J, Clauer U, et al. The efficacy of N-acetylcysteine as a hepatoprotective agent in liver transplantation[J]. Transpl Int, 1998, 11(S1): S390–S392. doi: 10.1111/j.1432-2277.1998.tb01164.x
|
| [91] |
Weigand MA, Plachky J, Thies JC, et al. N-acetylcysteine attenuates the increase in α-glutathione S-transferase and circulating ICAM-1 and VCAM-1 after reperfusion in humans undergoing liver transplantation[J]. Transplantation, 2001, 72(4): 694–698. doi: 10.1097/00007890-200108270-00023
|
| [92] |
Bucuvalas JC, Ryckman FC, Krug S, et al. Effect of treatment with prostaglandin E1 and N-acetylcysteine on pediatric liver transplant recipients: a single-center study[J]. Pediatr Transplant, 2001, 5(4): 274–278. doi: 10.1034/j.1399-3046.2001.005004274.x
|
| [93] |
Bromley PN, Cottam SJ, Hilmi I, et al. Effects of intraoperative N-acetylcysteine in orthotopic liver transplantation[J]. Br J Anaesth, 1995, 75(3): 352–354. doi: 10.1093/bja/75.3.352
|
| [94] |
Steib A, Freys G, Collin F, et al. Does N-acetylcysteine improve hemodynamics and graft function in liver transplantation?[J]. Liver Transpl Surg, 1998, 4(2): 152–157. doi: 10.1002/(ISSN)1527-6473a
|
| [95] |
Tsuchihashi SI, Fondevila C, Shaw GD, et al. Molecular characterization of rat leukocyte P-selectin glycoprotein ligand-1 and effect of its blockade: protection from ischemia-reperfusion injury in liver transplantation[J]. J Immunol, 2006, 176(1): 616–624. doi: 10.4049/jimmunol.176.1.616
|
| [96] |
Dulkanchainun TS, Goss JA, Imagawa DK, et al. Reduction of hepatic ischemia/reperfusion injury by a soluble P-selectin glycoprotein ligand-1[J]. Ann Surg, 1998, 227(6): 832–840. doi: 10.1097/00000658-199806000-00006
|
| [97] |
Amersi F, Farmer DG, Shaw GD, et al. P-selectin glycoprotein ligand-1 (rPSGL-Ig)-mediated blockade of CD62 selectin molecules protects rat steatotic liver grafts from ischemia/reperfusion injury[J]. Am J Transplant, 2002, 2(7): 600–608. doi: 10.1034/j.1600-6143.2002.20704.x
|
| [98] |
Busuttil RW, Lipshutz GS, Kupiec-Weglinski JW, et al. rPSGL-Ig for improvement of early liver allograft function: a double-blind, placebo-controlled, single-center phase II study[J]. Am J Transplant, 2011, 11(4): 786–797. doi: 10.1111/j.1600-6143.2011.03441.x
|
| [99] |
Valentino KL, Gutierrez M, Sanchez R, et al. First clinical trial of a novel caspase inhibitor: anti-apoptotic caspase inhibitor, IDN-6556, improves liver enzymes[J]. Int J Clin Pharmacol Ther, 2003, 41(10): 441–449. doi: 10.5414/CPP41441
|
| [100] |
Linton SD, Aja T, Armstrong RA, et al. First-in-class pan caspase inhibitor developed for the treatment of liver disease[J]. J Med Chem, 2005, 48(22): 6779–6782. doi: 10.1021/jm050307e
|
| [101] |
Baskin-Bey ES, Washburn K, Feng S, et al. Clinical trial of the pan-caspase inhibitor, IDN-6556, in human liver preservation injury[J]. Am J Transplant, 2007, 7(1): 218–225. doi: 10.1111/ajt.2007.7.issue-1
|
| [102] |
Song G, Ouyang GL, Bao SD. The activation of Akt/PKB signaling pathway and cell survival[J]. J Cell Mol Med, 2005, 9(1): 59–71. doi: 10.1111/jcmm.2005.9.issue-1
|
| [103] |
Covington SM, Bauler LD, Toledo-Pereyra LH. Akt: a therapeutic target in hepatic ischemia-reperfusion injury[J]. J Invest Surg, 2017, 30(1): 47–55. doi: 10.1080/08941939.2016.1206999
|
| [104] |
Koh PO. Melatonin prevents hepatic injury-induced decrease in Akt downstream targets phosphorylations[J]. J Pineal Res, 2011, 51(2): 214–219. doi: 10.1111/j.1600-079X.2011.00879.x
|
| [105] |
Bertoldo MJ, Faure M, Dupont J, et al. AMPK: a master energy regulator for gonadal function[J]. Front Neurosci, 2015, 9: 235.
|
| [106] |
Peralta C, Bartrons R, Serafin A, et al. Adenosine monophosphate-activated protein kinase mediates the protective effects of ischemic preconditioning on hepatic ischemia-reperfusion injury in the rat[J]. Hepatology, 2001, 34(6): 1164–1173. doi: 10.1053/jhep.2001.29197
|
| [107] |
Ding WX, Zhang Q, Dong YB, et al. Adiponectin protects the rats liver against chronic intermittent hypoxia induced injury through AMP-activated protein kinase pathway[J]. Sci Rep, 2016, 6: 34151. doi: 10.1038/srep34151
|
| [108] |
Zhang CZ, Liao Y, Li Q, et al. Recombinant adiponectin ameliorates liver ischemia reperfusion injury via activating the AMPK/eNOS pathway[J]. PLoS One, 2013, 8(6): e66382. doi: 10.1371/journal.pone.0066382
|
| [109] |
|
| [110] |
Marion-Letellier R, Savoye G, Ghosh S. Fatty acids, eicosanoids and PPAR gamma[J]. Eur J Pharmacol, 2016, 785: 44–49. doi: 10.1016/j.ejphar.2015.11.004
|
| [111] |
Zhou YL, Jia S, Wang CJ, et al. FAM3A is a target gene of peroxisome proliferator-activated receptor gamma[J]. Biochim Biophys Acta, 2013, 1830(8): 4160–4170. doi: 10.1016/j.bbagen.2013.03.029
|
| [112] |
Yang WL, Chen J, Meng YH, et al. Novel targets for treating ischemia-reperfusion injury in the liver[J]. Int J Mol Sci, 2018, 19(5): E1302. doi: 10.3390/ijms19051302
|
| [113] |
Xu CF, Yu CH, Li YM. Regulation of hepatic microRNA expression in response to ischemic preconditioning following ischemia/reperfusion injury in mice[J]. OMICS, 2009, 13(6): 513–520. doi: 10.1089/omi.2009.0035
|
| [114] |
Gehrau RC, Mas VR, Dumur CI, et al. Regulation of molecular pathways in ischemia-reperfusion injury after liver transplantation[J]. Transplantation, 2013, 96(10): 926–934. doi: 10.1097/TP.0b013e3182a20398
|
| [115] |
Mard SA, Akbari G, Dianat M, et al. Protective effects of crocin and zinc sulfate on hepatic ischemia-reperfusion injury in rats: a comparative experimental model study[J]. Biomed Pharmacother, 2017, 96: 48–55. doi: 10.1016/j.biopha.2017.09.123
|
| [116] |
Peralta C, Hotter G, Closa D, et al. Protective effect of preconditioning on the injury associated to hepatic ischemia-reperfusion in the rat: role of nitric oxide and adenosine[J]. Hepatology, 1997, 25(4): 934–937. doi: 10.1002/hep.510250424
|
| [117] |
Quarrie R, Cramer BM, Lee DS, et al. Ischemic preconditioning decreases mitochondrial proton leak and reactive oxygen species production in the postischemic heart[J]. J Surg Res, 2011, 165(1): 5–14. doi: 10.1016/j.jss.2010.09.018
|
| [118] |
Richards JA, Wigmore SJ, Devey LR. Heme oxygenase system in hepatic ischemia-reperfusion injury[J]. World J Gastroenterol, 2010, 16(48): 6068–6078. doi: 10.3748/wjg.v16.i48.6068
|
| [119] |
Liu AD, Fang HS, Wei WW, et al. Ischemic preconditioning protects against liver ischemia/reperfusion injury via heme oxygenase-1-mediated autophagy[J]. Crit Care Med, 2014, 42(12): e762–e771. doi: 10.1097/CCM.0000000000000659
|
| [120] |
Rüdiger HA, Graf R, Clavien PA. Sub-lethal oxidative stress triggers the protective effects of ischemic preconditioning in the mouse liver[J]. J Hepatol, 2003, 39(6): 972–977. doi: 10.1016/S0168-8278(03)00415-X
|
| [121] |
Rolo AP, Teodoro JS, Peralta C, et al. Prevention of I/R injury in fatty livers by ischemic preconditioning is associated with increased mitochondrial tolerance: the key role of ATPsynthase and mitochondrial permeability transition[J]. Transpl Int, 2009, 22(11): 1081–1090. doi: 10.1111/tri.2009.22.issue-11
|
| [122] |
Abu-Amara M, Yang SY, Quaglia A, et al. Role of endothelial nitric oxide synthase in remote ischemic preconditioning of the mouse liver[J]. Liver Transpl, 2011, 17(5): 610–619. doi: 10.1002/lt.v17.5
|
| [123] |
Koti RS, Seifalian AM, Davidson BR. Protection of the liver by ischemic preconditioning: a review of mechanisms and clinical applications[J]. Dig Surg, 2003, 20(5): 383–396. doi: 10.1159/000072064
|
| [124] |
Gurusamy KS, Kumar Y, Sharma D, et al. Ischaemic preconditioning for liver transplantation[J]. Cochrane Database Syst Rev, 2008, (1): CD006315.
|
| [125] |
Nadarajah L, Yaqoob MM, McCafferty K. Ischemic conditioning in solid organ transplantation: is it worth giving your right arm for?[J]. Curr Opin Nephrol Hypertens, 2017, 26(6): 467–476. doi: 10.1097/MNH.0000000000000367
|
| [126] |
Koneru B, Fisher A, He Y, et al. Ischemic preconditioning in deceased donor liver transplantation: a prospective randomized clinical trial of safety and efficacy[J]. Liver Transpl, 2005, 11(2): 196–202. doi: 10.1002/(ISSN)1527-6473
|
| [127] |
Koneru B, Shareef A, Dikdan G, et al. The ischemic preconditioning paradox in deceased donor liver transplantation-evidence from a prospective randomized single blind clinical trial[J]. Am J Transplant, 2007, 7(12): 2788–2796. doi: 10.1111/ajt.2007.7.issue-12
|
| [128] |
Theodoraki K, Karmaniolou I, Tympa A, et al. Beyond preconditioning: postconditioning as an alternative technique in the prevention of liver ischemia-reperfusion injury[J]. Oxid Med Cell Longev, 2016, 2016: 8235921.
|
| [129] |
Sun K, Liu ZS, Sun Q. Role of mitochondria in cell apoptosis during hepatic ischemia-reperfusion injury and protective effect of ischemic postconditioning[J]. World J Gastroenterol, 2004, 10(13): 1934–1938. doi: 10.3748/wjg.v10.i13.1934
|
| [130] |
Zhang WX, Yin W, Zhang L, et al. Preconditioning and postconditioning reduce hepatic ischemia-reperfusion injury in rats[J]. Hepatobiliary Pancreat Dis Int, 2009, 8(6): 586–590.
|
| [131] |
Yoon SY, Kim CY, Han HJ, et al. Protective effect of ischemic postconditioning against hepatic ischemic reperfusion injury in rat liver[J]. Ann Surg Treat Res, 2015, 88(5): 241–245. doi: 10.4174/astr.2015.88.5.241
|
| [132] |
Lin HC, Lee TK, Tsai CC, et al. Ischemic postconditioning protects liver from ischemia-reperfusion injury by modulating mitochondrial permeability transition[J]. Transplantation, 2012, 93(3): 265–271. doi: 10.1097/TP.0b013e31823ef335
|
| [133] |
Wang N, Lu JG, He XL, et al. Effects of ischemic postconditioning on reperfusion injury in rat liver grafts after orthotopic liver transplantation[J]. Hepatol Res, 2009, 39(4): 382–390. doi: 10.1111/hep.2009.39.issue-4
|
| [134] |
Kim WH, Lee JH, Ko JS, et al. Effect of remote ischemic postconditioning on patients undergoing living donor liver transplantation[J]. Liver Transpl, 2014, 20(11): 1383–1392. doi: 10.1002/lt.23960
|
| [135] |
Ricca L, Lemoine A, Cauchy F, et al. Ischemic postconditioning of the liver graft in adult liver transplantation[J]. Transplantation, 2015, 99(8): 1633–1643. doi: 10.1097/TP.0000000000000685
|
| [136] |
Schlegel AA, Kalisvaart M, Muiesan P. Machine perfusion in liver transplantation: an essential treatment or just an expensive toy?[J]. Minerva Anestesiol, 2018, 84(2): 236–245.
|
| [137] |
Liu Q, Vekemans K, Iania L, et al. Assessing warm ischemic injury of pig livers at hypothermic machine perfusion[J]. J Surg Res, 2014, 186(1): 379–389. doi: 10.1016/j.jss.2013.07.034
|
| [138] |
Monbaliu D, Liu Q, Libbrecht L, et al. Preserving the morphology and evaluating the quality of liver grafts by hypothermic machine perfusion: a proof-of-concept study using discarded human livers[J]. Liver Transpl, 2012, 18(12): 1495–1507. doi: 10.1002/lt.v18.12
|
| [139] |
Manekeller S, Schuppius A, Stegemann J, et al. Role of perfusion medium, oxygen and rheology for endoplasmic reticulum stress-induced cell death after hypothermic machine preservation of the liver[J]. Transpl Int, 2008, 21(2): 169–177.
|
| [140] |
Jain S, Xu HZ, Duncan H, et al. Ex-vivo study of flow dynamics and endothelial cell structure during extended hypothermic machine perfusion preservation of livers[J]. Cryobiology, 2004, 48(3): 322–332. doi: 10.1016/j.cryobiol.2004.01.010
|
| [141] |
Schlegel A, de Rougemont O, Graf R, et al. Protective mechanisms of end-ischemic cold machine perfusion in DCD liver grafts[J]. J Hepatol, 2013, 58(2): 278–286. doi: 10.1016/j.jhep.2012.10.004
|
| [142] |
Gallinat A, Efferz P, Paul A, et al. One or 4 h of " in-house” reconditioning by machine perfusion after cold storage improve reperfusion parameters in porcine kidneys[J]. Transpl Int, 2014, 27(11): 1214–1219. doi: 10.1111/tri.2014.27.issue-11
|
| [143] |
Guarrera JV, Henry SD, Samstein B, et al. Hypothermic machine preservation facilitates successful transplantation of " orphan” extended criteria donor livers[J]. Am J Transplant, 2015, 15(1): 161–169. doi: 10.1111/ajt.12958
|
| [144] |
Dutkowski P, Schlegel A, de Oliveira M, et al. HOPE for human liver grafts obtained from donors after cardiac death[J]. J Hepatol, 2014, 60(4): 765–772. doi: 10.1016/j.jhep.2013.11.023
|
| [145] |
Schlegel A, Muller X, Kalisvaart M, et al. Outcomes of DCD liver transplantation using organs treated by hypothermic oxygenated perfusion before implantation[J]. J Hepatol, 2019, 70(1): 50–57.
|
| [146] |
Ravikumar R, Jassem W, Mergental H, et al. Liver transplantation after ex vivo normothermic machine preservation: a phase 1 (first-in-man) clinical trial[J]. Am J Transplant, 2016, 16(6): 1779–1787. doi: 10.1111/ajt.13708
|
| [147] |
Xu HZ, Berendsen T, Kim K, et al. Excorporeal normothermic machine perfusion resuscitates pig DCD livers with extended warm ischemia[J]. J Surg Res, 2012, 173(2): e83–e88. doi: 10.1016/j.jss.2011.09.057
|
| [148] |
Mergental H, Perera MTPR, Laing RW, et al. Transplantation of declined liver allografts following normothermic ex-situ evaluation[J]. Am J Transplant, 2016, 16(11): 3235–3245. doi: 10.1111/ajt.13875
|
| [149] |
Jassem W, Xystrakis E, Ghnewa YG, et al. Normothermic machine perfusion (NMP) inhibits proinflammatory responses in the liver and promotes regeneration[J]. Hepatology, 2018. doi: 10.1002/hep.30475[Epub ahead of print
|
| [150] |
Balfoussia D, Yerrakalva D, Hamaoui K, et al. Advances in machine perfusion graft viability assessment in kidney, liver, pancreas, lung, and heart transplant[J]. Exp Clin Transplant, 2012, 10(2): 87–100. doi: 10.6002/ect
|
| [151] |
Watson CJE, Randle LV, Kosmoliaptsis V, et al. 26-hour storage of a declined liver before successful transplantation using ex vivo normothermic perfusion[J]. Ann Surg, 2017, 265(1): e1–e2. doi: 10.1097/SLA.0000000000001834
|
| [152] |
Laing RW, Bhogal RH, Wallace L, et al. The use of an acellular oxygen carrier in a human liver model of normothermic machine perfusion[J]. Transplantation, 2017, 101(11): 2746–2756. doi: 10.1097/TP.0000000000001821
|
| [153] |
op den Dries S, Karimian N, Sutton ME, et al. Ex vivo normothermic machine perfusion and viability testing of discarded human donor livers[J]. Am J Transplant, 2013, 13(5): 1327–1335. doi: 10.1111/ajt.12187
|
| [154] |
Braat AE, Blok JJ, Putter H, et al. The eurotransplant donor risk index in liver transplantation: ET-DRI[J]. Am J Transplant, 2012, 12(10): 2789–2796. doi: 10.1111/j.1600-6143.2012.04195.x
|
| [155] |
Feng S, Goodrich NP, Bragg-Gresham JL, et al. Characteristics associated with liver graft failure: the concept of a donor risk index[J]. Am J Transplant, 2006, 6(4): 783–790. doi: 10.1111/j.1600-6143.2006.01242.x
|
| [156] |
Perera T, Mergental H, Stephenson B, et al. First human liver transplantation using a marginal allograft resuscitated by normothermic machine perfusion[J]. Liver Transpl, 2016, 22(1): 120–124. doi: 10.1002/lt.24369
|
| [157] |
Watson CJE, Kosmoliaptsis V, Randle LV, et al. Normothermic perfusion in the assessment and preservation of declined livers before transplantation: hyperoxia and vasoplegia-important lessons from the first 12 cases[J]. Transplantation, 2017, 101(5): 1084–1098. doi: 10.1097/TP.0000000000001661
|
| [158] |
Khorsandi SE, Quaglia A, Salehi S, et al. The microRNA expression profile in donation after cardiac death (DCD) livers and its ability to identify primary non function[J]. PLoS One, 2015, 10(5): e0127073. doi: 10.1371/journal.pone.0127073
|
| [159] |
Bruinsma BG, Sridharan GV, Weeder PD, et al. Metabolic profiling during ex vivo machine perfusion of the human liver[J]. Sci Rep, 2016, 6: 22415. doi: 10.1038/srep22415
|
| [160] |
Nasralla D, Coussios CC, Mergental H, et al. A randomized trial of normothermic preservation in liver transplantation[J]. Nature, 2018, 557(7703): 50–56. doi: 10.1038/s41586-018-0047-9
|
| [161] |
Durand F, Renz JF, Alkofer B, et al. Report of the Paris consensus meeting on expanded criteria donors in liver transplantation[J]. Liver Transpl, 2008, 14(12): 1694–1707. doi: 10.1002/lt.v14:12
|
| [162] |
Spitzer AL, Lao OB, Dick AAS, et al. The biopsied donor liver: incorporating macrosteatosis into high-risk donor assessment[J]. Liver Transpl, 2010, 16(7): 874–884. doi: 10.1002/lt.v16:7
|
| [163] |
Nativ NI, Maguire TJ, Yarmush G, et al. Liver defatting: an alternative approach to enable steatotic liver transplantation[J]. Am J Transplant, 2012, 12(12): 3176–3183. doi: 10.1111/ajt.2012.12.issue-12
|
| [164] |
Nagrath D, Xu HZ, Tanimura Y, et al. Metabolic preconditioning of donor organs: defatting fatty livers by normothermic perfusion ex vivo[J]. Metab Eng, 2009, 11(4–5): 274–283. doi: 10.1016/j.ymben.2009.05.005
|
| [165] |
Boteon YL, Afford SC, Mergental H. Pushing the limits: machine preservation of the liver as a tool to recondition high-risk grafts[J]. Curr Transplant Rep, 2018, 5(2): 113–120. doi: 10.1007/s40472-018-0188-7
|
| [166] |
Goldaracena N, Echeverri J, Spetzler VN, et al. Anti-inflammatory signaling during ex vivo liver perfusion improves the preservation of pig liver grafts before transplantation[J]. Liver Transpl, 2016, 22(11): 1573–1583. doi: 10.1002/lt.v22.11
|
| [167] |
Morales-Ruiz M, Fondevila C, Muñoz-Luque J, et al. Gene transduction of an active mutant of akt exerts cytoprotection and reduces graft injury after liver transplantation[J]. Am J Transplant, 2007, 7(4): 769–778. doi: 10.1111/ajt.2007.7.issue-4
|
| [168] |
Van Raemdonck D, Neyrinck A, Rega F, et al. Machine perfusion in organ transplantation: a tool for ex-vivo graft conditioning with mesenchymal stem cells?[J]. Curr Opin Organ Transplant, 2013, 18(1): 24–33. doi: 10.1097/MOT.0b013e32835c494f
|
 Authors and Reviewers
Authors and Reviewers

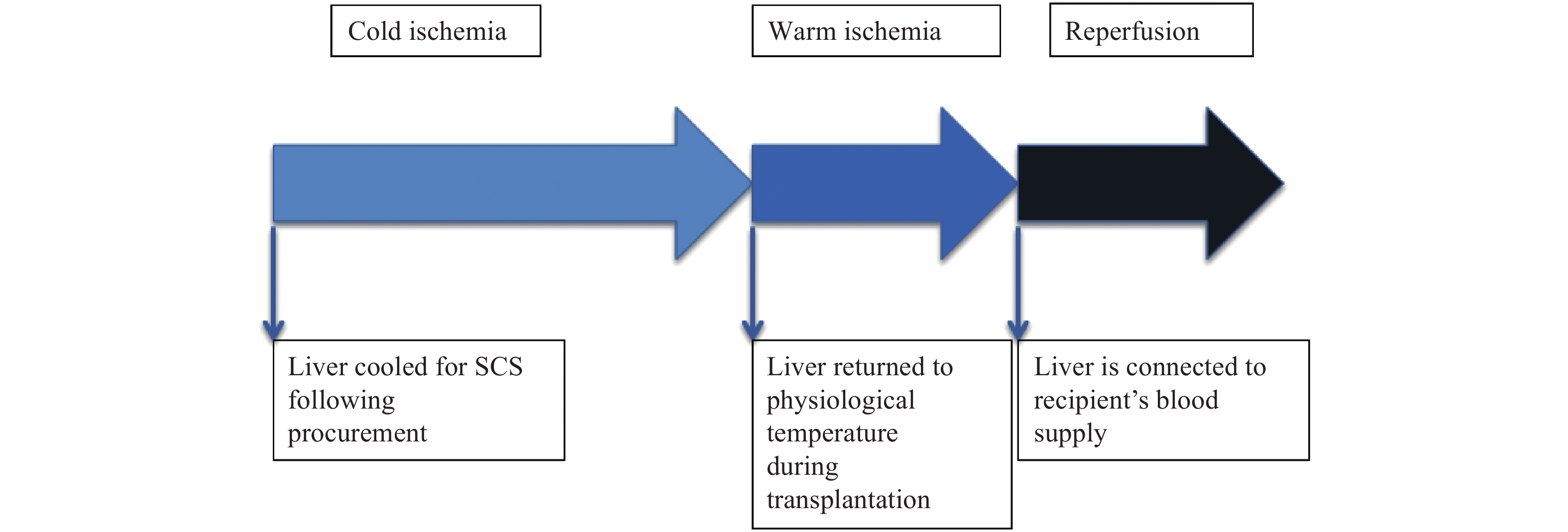
 DownLoad:
DownLoad: