Anastasia V. Poznyak, Alexey Aleksandrovich Yakovlev, Mikhail А. Popov, Alexander D. Zhuravlev, Vasily N. Sukhorukov, Alexander N. Orekhov. Coronary atherosclerotic plaque regression strategies[J]. The Journal of Biomedical Research. DOI: 10.7555/JBR.37.20230223
Citation:
Anastasia V. Poznyak, Alexey Aleksandrovich Yakovlev, Mikhail А. Popov, Alexander D. Zhuravlev, Vasily N. Sukhorukov, Alexander N. Orekhov. Coronary atherosclerotic plaque regression strategies[J]. The Journal of Biomedical Research. DOI: 10.7555/JBR.37.20230223
Anastasia V. Poznyak, Alexey Aleksandrovich Yakovlev, Mikhail А. Popov, Alexander D. Zhuravlev, Vasily N. Sukhorukov, Alexander N. Orekhov. Coronary atherosclerotic plaque regression strategies[J]. The Journal of Biomedical Research. DOI: 10.7555/JBR.37.20230223
Citation:
Anastasia V. Poznyak, Alexey Aleksandrovich Yakovlev, Mikhail А. Popov, Alexander D. Zhuravlev, Vasily N. Sukhorukov, Alexander N. Orekhov. Coronary atherosclerotic plaque regression strategies[J]. The Journal of Biomedical Research. DOI: 10.7555/JBR.37.20230223
Unproofed Manuscript: The manuscript has been professionally copyedited and typeset to confirm the JBR’s formatting, but still needs proofreading by the corresponding author to ensure accuracy and correct any potential errors introduced during the editing process. It will be replaced by the online publication version.
Anastasia V. Poznyak, Institute for Atherosclerosis Research, Osennyaya Street 4-1-207, Moscow121609, Russia. E-mail: tehhy_85@mail.ru
Alexander N. Orekhov, Laboratory of Angiopathology, Institute of General Pathology and Pathophysiology, Moscow 125315, Russia. E-mail: alexandernikolaevichorekhov@gmail.com
Atherosclerosis poses a significant and widespread problem at the population level. Consequently, there is a pressing need to develop effective methods to reduce the risk associated with this condition, which holds a prominent position in cardiology research. The primary manifestation of atherosclerosis involves plaque formation on the walls of coronary arteries. These plaques not only disrupt blood flow but also raise the likelihood of thrombosis and subsequent cardiovascular events. Unfortunately, atherosclerosis itself is usually asymptomatic, resulting in challenges with diagnosis and a delayed initiation of treatment. Hence, strategies focusing on the regression of existing plaques within blood vessels play a crucial role. Our review encompasses comprehensive data on the regression of coronary atherosclerotic plaques, examining both the underlying mechanisms and a range of regression strategies, encompassing lifestyle modifications to medical interventions.
Atherosclerosis, a complex disease characterized by the formation of plaques within the large arteries, remains a significant global health challenge[1]. These plaques consist of fatty deposits, inflammatory cells, smooth muscle cells, and extracellular matrix components[2]. The progressive arterial narrowing caused by these plaques compromises blood flow to the heart muscle, potentially leading to life-threatening cardiovascular events[3]. While extensive investigations have focused on understanding the progression of coronary atherosclerosis, recent studies have brought attention to a captivating phenomenon: plaque regression, referring to a favorable process in which atherosclerotic plaques, either partially or fully, shrink in size or become more stable[4]. This regression enhances blood flow and reduces vulnerability to plaque rupture, holding tremendous promise for the development of innovative strategies in the prevention and treatment of coronary artery disease[4]. However, the understanding of plaque regression is still evolving, and controversies and concerns surround this topic. One area of debate is the mechanisms underlying plaque regression. Investigators are actively exploring the roles of various factors, including modifications in lipid profiles, inflammation resolution, extracellular matrix remodeling, and neovascularization, in driving the regression process. Understanding the interplay among these factors and their contribution to plaque regression remains an ongoing research endeavor. Another aspect of concern is clinical implications of plaque regression. While the regression of atherosclerotic plaques is generally considered beneficial, questions arise regarding how to assess the occurrence and extent of regression accurately. The development of reliable imaging techniques and biomarkers to monitor plaque regression is a crucial area of investigation. Furthermore, there are gaps in our knowledge regarding the long-term outcomes and sustainability of plaque regression. Additional investigations are needed to determine the effect of plaque regression on clinical outcomes, such as the improved prognosis and the reduced incidence of cardiovascular events, which will help elucidate the role of plaque regression in guiding clinical management and risk stratification of patients with coronary artery disease.
In summary, although plaque regression holds a great promise as a therapeutic target and a marker of the improved cardiovascular health, there are ongoing research efforts to better understand its underlying mechanisms, develop accurate assessment methods, and determine its long-term clinical implications. By addressing these research gaps and controversies, we can advance our knowledge and pave the way for more effective strategies in the prevention and treatment of coronary artery disease. Ultimately, our review aims to consolidate the existing evidence surrounding plaque regression in coronary atherosclerosis. By uncovering the intricate molecular and cellular processes involved in plaque regression, we can gain valuable insights into potential therapeutic targets and interventions to mitigate the burden of atherosclerosis.
Mechanism of atherosclerosis reversal and regression
Since the beginning of the 20th century, the developer of medical therapy has proposed the possibility of atherosclerosis regression through lipid level reduction. Experimental studies have been conducted to examine the influence of diet on plaque regression. The results showed a reduction in plaque area in animal models by transitioning from a high-cholesterol diet to a low-cholesterol diet, suggesting the potential for plaque regression[5]. According to Brown et al[6], the macrophage content can be swiftly corrected through the plaque lipoprotein environment; this correction leads to a decrease in the number of foam cells and intraplaque macrophages, a downregulation of inflammatory markers, and an increase in anti-inflammatory markers.
A large decrease in levels of apolipoprotein B-containing lipoproteins in plasma, coupled with an increase in lipid efflux from the plaque, causes significant improvements in plaque microenvironments, coinciding with lesion regression[7]. The increased emigration of macrophages from the plaque leads to a gradual decrease in their numbers, with a reduction of extracellular cholesterol volume and foam cells, and an involvement of anti-inflammatory phagocytes in necrotic tissue removal, and their regeneration to replace inflammatory macrophages, which are essential parts of the complex process of plaque shrinkage[7].
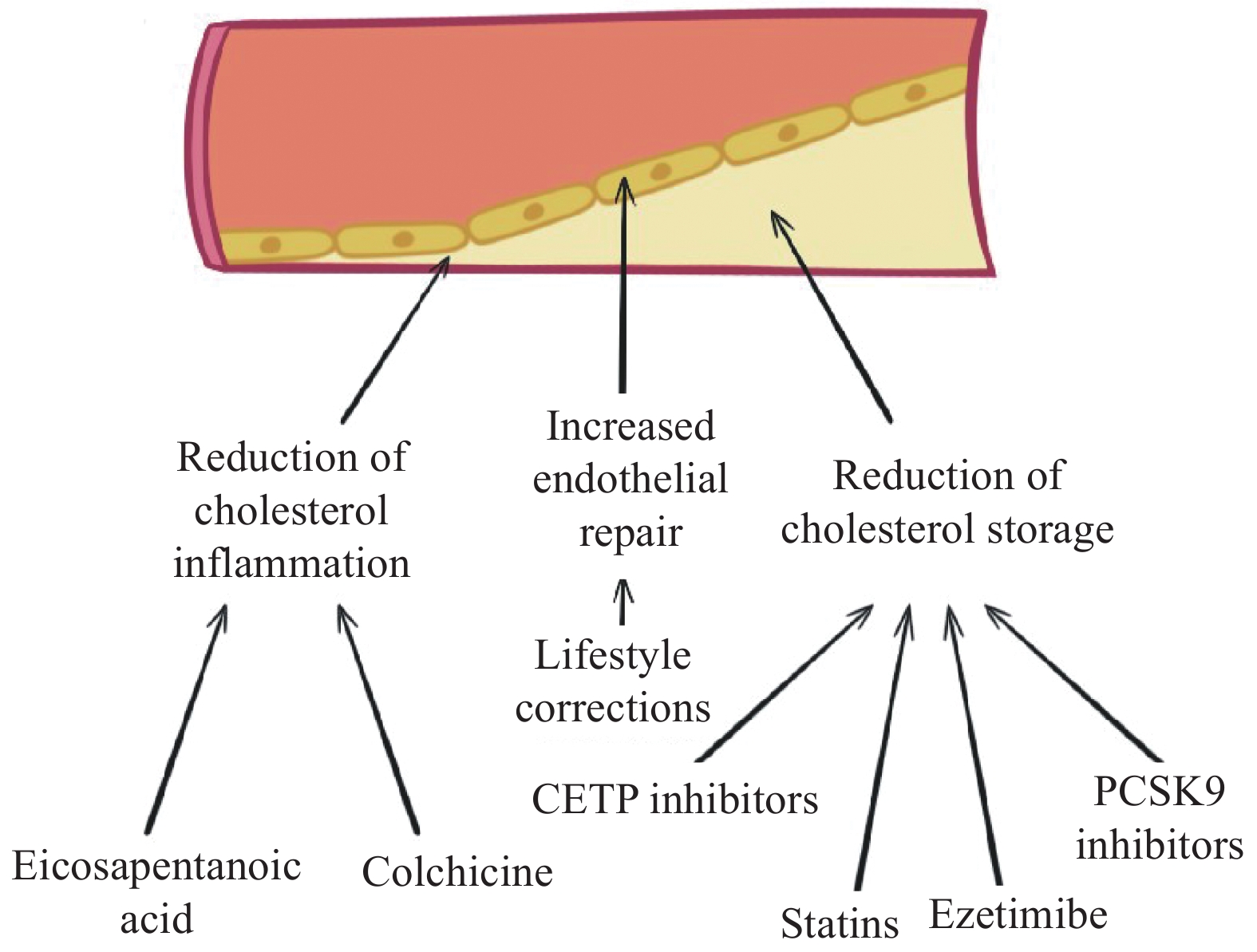
Therefore, the notable achievements in the utilization of statins, fibrates, and other hypolipidemic agents in clinical studies are primarily attributed to the induction of stability in small vulnerable plaques that include thick clusters of intimal macrophages and a large lipid core[8]. Despite representing only about 20% of the total number of plaques, these lesions are responsible for approximately 85% of severe clinical events[8]. Figure 1 shows the variety of strategies aimed at coronary plaque regression.
Figure
1.
Therapeutic strategies targeting three main aspects of plaque progression.
There are several main strategies of plaque regression: reduction of inflammation (released through colchicine, eicosopentanoic acid), endothelial repair (achieved via lifestyle correction), and reduction of the cholesterol storage (CETP inhibitors, statins, ezetimibe, PCSK9 inhibitors).
Plaque regression, the reduction in the size and composition of atherosclerotic plaques, can occur due to several possible mechanisms. (1) Intensive lipid-lowering therapies, such as statins, play a crucial role in reducing low-density lipoprotein (LDL) cholesterol levels, which in turn decreases the supply of lipids available for deposition within the plaque and leads to its regression[9]; (2) Atherosclerosis involves chronic inflammation within the arterial wall, and medications targeting inflammation, such as high-dose statins and anti-inflammatory drugs, can effectively suppress inflammation, stabilize plaques, and promote regression[10]; (3) Smooth muscle cell migration is another important aspect of plaque regression, and drug interventions like the statin therapy and angiotensin-converting enzyme inhibitors facilitate the migration of smooth muscle cells from the plaque region, resulting in plaque regression[11]; (4) Furthermore, plaque regression may involve changes in plaque calcification, and there have been explorations into treatments like sodium thiosulfate that aim to promote the dissolution of calcified deposits within the plaque, thus facilitating regression[12]; and (5) additionally, promoting an increased plaque stability is crucial in reducing the risk of plaque rpture and subsequent cardiovascular events. Medications like statins improve plaque stability by decreasing inflammation, promoting fibrous cap thickening, and inhibiting the degradation of extracellular matrix proteins[13]. It is important to acknowledge that the mechanisms involved in plaque regression are complex and multifactorial, and ongoing research is needed to gain a better understanding and optimize therapies aimed at promoting plaque regression.
Animal models
Rabbit models for coronary plaque regression
Studies investigating coronary plaque regression have been conducted using rabbit models, which provide valuable insights into the underlying mechanisms and potential treatments[14]. These animal models offer several advantages, including a similar lipid profile to humans, rapid plaque development, and the ability to manipulate genetic factors[15]. Investigators have used various interventions, such as cholesterol-lowering drugs, anti-inflammatory agents, and gene therapies, to induce plaque regression in rabbits[15]. Through histopathological analysis and imaging techniques, these studies have demonstrated a decrease in plaque size, a reversal of lipid deposition, and an increase in smooth muscle cell content after interventions[16]. Additionally, studies have elucidated key molecular pathways involved in plaque regression, including the modulation of lipoprotein metabolism, reduction in inflammation, and enhancement of cholesterol efflux[17]. Rabbit models continue to play a crucial role in understanding the complex process of plaque regression and evaluating novel therapeutic strategies for the treatment of atherosclerosis[18].
Murine models of atherosclerosis regression
Compared with baseline plaques, the regression of atherosclerotic cardiovascular disease in mice encompasses at least one of the following: a decrease in plaque size, macrophage content, or cholesterol levels[19]. All murine models of atherosclerosis regression share two crucial phases, including a beginning progression phase to establish a baseline plaque with a high level of plasma apolipoprotein B-containing LDL-C concentration above 300 mg/dL [19], and a subsequent phase to involve low levels of atherogenic lipoproteins, such as LDL cholesterol and very-LDL, to lead to atherosclerosis regression[20]. Preclinical murine models for the regression of atherosclerotic cardiovascular disease are considered analogous to a high-intensity statin therapy in humans[20].
Plaque transplantation
In the first mouse model of atherosclerosis regression, plaque transplantation was performed by transplanting thoracic arch arteries or segments of pharyngeal arch arteries with atherosclerotic lesions from mice donors with hyperlipidemia (e.g., ApoE−/− or Ldlr−/−) into recipient mice with normolipemia (i.e., C57Bl/6J)[21]. As a result, a decreased lipid area, a reduced macrophage presence, and a decline in lesions were observed in recipient mice, indicating the plaque regression caused by rapid changes in apolipoprotein B. This model of plaque transplantation provides crucial data revealing the mechanisms of plaque regression and evaluating the treatment of atherosclerotic cardiovascular disease.
Elimination of atherogenic lipoprotein production
A reversible atherosclerosis model has been represented by Reversa mice that are genetically modified with a conditional allele of microsomal triglyceride transfer protein (Mttp) introduced into the liver (Ldlr−/−Apob100/100Mttpfl/flMx1-Cre+/+) and a lack of LDL receptors[22]. By inhibiting MTTP expression, such as with interferon-alpha (IFN-α), IFN-β, or synthetic double-stranded RNA (e.g., polyinosinic-polycytidylic acid, pIpC), in combination with a change to a chow diet, a fast plaque regression and a reduction in very-LDL and LDL, along with a decrease in apolipoprotein B cholesterol levels from over 1000 mg/dL to less than 150 mg/dL, were achieved[22]. Similar results in plaque regression and transformation of atherogenic lipoproteins were observed in Ldlr−/− mice with MTTP inhibition using BMS 212122[22]. Additionally, the regression of atherosclerotic cardiovascular disease in Ldlr−/− mice was induced by suppressing apoB production with an apolipoprotein B antisense oligonucleotide (ASO)[22].
Adeno-associated virus therapy for atherosclerosis regression
Adeno-associated virus has been previously used in therapy to decrease lipid levels in mice by inducing hepatic overexpression of the LDL receptor in Ldlr−/− mice and apolipoprotein E in ApoE−/− mice[23]. The concentration of LDL cholesterol is increased because of higher levels of PCSK9 (proprotein convertase subtilisin/kexin type 9), which degrades hepatic receptors for low-density lipoprotein, thereby causing atherosclerosis in the wild-type mice[24]. Thus, the PCSK9 adeno-associated virus model is used to induce regression by switching to a chow diet in animal studies, and the addition of an MTTP inhibitor to the diet accelerates the decrease in lipid levels[25]. The adeno-associated virus therapy, as one of the genotype-independent atherosclerosis models, facilitates a rapid evaluation of factors regulating regression in atherosclerotic cardiovascular disease, because it eliminates the need for complex and demanding backcrosses[25]. Moreover, it helps a quick induction of phenotypes specified by genes in adult mice with the established lesions facilitates mechanistic work to examine candidates capable of controlling regression[26].
Reduction in dietary cholesterol
In certain mouse models, mice prone to atherosclerosis with a high-fat diet show the regression of atherosclerosis when switched to a low-fat diet, which is compared with the effects of switching to a healthy diet or receiving statin treatment in humans[27]. However, the regression speed caused by this method is slower, and some mice may not show signs of regression because of insufficient normalization of apolipoprotein B cholesterol levels (e.g., ApoE−/− mice)[27].
Strategies to achieve plaque regression stabilization
Pharmacological treatments and dietary corrections with physical activity are fundamental strategies in plaque regression, though pharmacology has been by far more effective[28].
Physical activity
It is difficult to overestimate the impact of physical activity on the prevention of cardiovascular diseases. The influence of cardiac rehabilitation based on physical training on the reduction in the risk of hospitalization and cardiovascular mortality is 18% and 26%, respectively; these numbers were recently published in a Cochrane review and meta-analysis that included at least 63 studies[29], in which the effect of intense physical activity on plaque regression has been estimated in two randomized trials. The results obtained in both trials, by using intravascular ultrasound (IVUS) echocardiography, demonstrated no significant differences between treatment groups despite the registered plaque regression in each exercise arm [31]. Nevertheless, in a post hoc analysis of one of the trials, the evidence of differences in plaque regression levels was found between patients with different numbers of everyday walking steps. The participants who walked less than 7,000 steps per day had a lower percentage of plaque regression, compared with those who managed to walk at least 7,000 steps per day (<3.6% vs 12.5%; P < 0.05)[30]. Even though there is not much data on the correlation between plaque regression and exercise, one study managed to find evidence of the effect of physical activities on total atheroma volumes (TAVs)[31]. Using a multivariable analysis, the study demonstrated the connection between a score of lifestyle modification practices, consisting of BMI, smoking history, and exercise frequency, and TAV reduction over six months measured by ultrasound echocardiography. According to cross-sectional studies dedicated to the assessment of plaque composition among athletes, participants with a sedentary lifestyle had a higher risk of cardiac problems compared with athletes; moreover, cardiac CT angiography showed that participants with a low physical activity had greater mixed plaque morphologies, while athletes had a higher calcific plaque volume[31]. All of the aforementioned data have made an important contribution to the popularization of physical activity and exercise as tools for cardiovascular (CV) risk prevention and plaque regression.
Plaque calcification, a process characterized by the deposition of calcium within atherosclerotic plaques, has been a subject of interest and investigation in cardiovascular research[32]. The paradox observed with statins, a class of drugs commonly used to reduce cholesterol and stabilize plaques, lies in their potential to promote both regression and calcification of plaques[33]. Statins effectively lower LDL cholesterol levels, reduce inflammation, and improve plaque stability, which generally lead to plaque regression[34]. However, in some cases, statins may also enhance the deposition of calcium within the plaques, increasing their calcification[35]. The exact mechanism underlying this paradoxical effect is not fully understood. It is speculated that statins may influence the cellular and molecular processes involved in plaque calcification, such as the regulation of osteogenic differentiation of vascular smooth muscle cells[36]. Further investigation is necessary to unravel the complex interplay between statins, plaque regression, and calcification, to optimize therapeutic interventions and minimize potential paradoxical effects.
Diet
A crucial factor in cardiovascular risk is unhealthy dietary habits that include a calorie surplus leading to obesity, as well as consuming processed food and a large amount of sugar. Nevertheless, some controversial data advocates the limitation of the influence of diet interventions on plaque regression. During the long-term follow-up of Dietary Intervention to Stop Coronary Atherosclerosis in Computed Tomography study (DISCO-CT2), 92 patients were randomized into two groups: an experimental group, in which patients receiving a combination of optimal medical therapy (OMT) and systematic follow-up by a dietitian to adhere to the Dietary Approaches to Stop Hypertension (DASH) nutrition model, and a control group, in which patients receiving OMT alone[37]. The study results demonstrated a greater reduction in noncalcified plaque volumes determined by CT in the experimental group, compared with the control group, yet both groups showed a similar percent atheroma volume[37]. According to the study by Ornish et al[38] in 1998, it is possible to achieve a reduction in coronary atherosclerosis progression through vast interventions in diet and lifestyle; in which forty-eight patients, who were randomized into aerobic activities, smoking cessation, a 10% whole food vegetarian diet, and psychosocial support groups, demonstrated plaque regression determined by coronary angiography at the end of five years. It can be concluded that the effectiveness of dietary interventions alone for patients with coronary diseases has some limitations, as there is evidence of plaque volume progression for the majority of patients receiving statin treatment together with dietary approaches.
Smoking and alcohol
It is a well-known fact that smoking cigarettes can have serious consequences for health, even former smoking is attributed to a high risk of subclinical atherosclerosis, ischemic events, and coronary calcification. Moreover, after smoking cessation, indexes of atherosclerotic burden decrease slowly. Results obtained in two cross-sectional studies showed an increase in coronary artery plaque burden, necrotic core volumes, and fibro-fatty volume with a reduced fibrous plaque volume in smokers compared to non-smoking individuals, as observed using IVUS; however, there were no signs of differences in calcification or cap thickness[39]. Because no longitudinal surveys have been conducted, the effect of smoking cessation on coronary plaque burden is still unknown.
There are not much data about the effect of alcohol on plaque progression either. A cross-sectional study showed that the levels of macrophage infiltration and lipid core volume among subjects who underwent femoral endarterectomy as well as coronary calcification measured by CT, were both inversely associated with alcohol consumption[40]. However, no studies providing visualization of coronary plaque volumes or longitudinal studies have been conducted[41].
Pharmacological treatments
Statins
Statins have been the cornerstone of plaque regression treatment since the beginning of the 1990s. Multiple meta-analyses and trials have shown the effectiveness of statins in decreasing cases of ischemic events and fatal outcomes for patients at risk of, or with established, cardiovascular diseases[42]. As the HMGCR protein inhibitors, statins successfully reduce LDL cholesterol levels by 30%–50%, and LDL cholesterol is commonly used to assess treatment response, given the correlation between a reduced risk of ischemic events and a potential reduction of LDL cholesterol levels[43].
Numerous studies have examined statins and their effect on plaque regression. Despite the heterogeneity of these studies, their findings have generally demonstrated the ability of statins to induce plaque regression. When compared with control groups with an approximately 10% progression of TAV, statins have led to a reduction in TAV ranging from 0% to 20%[44]. Most of these studies use intravascular echocardiography to evaluate the extent of plaque burden. Several studies employ coronary CT angiography, while other techniques, such as virtual histology IVUS, integrated backscatter IVUS, optical coherence tomography (OCT), near-infrared spectroscopy (NIRS), or fluorodeoxyglucose positron emission tomography, are also used to assess plaque composition[44–45].
IVUS provides high-resolution cross-sectional images of the vessel wall, allowing for a detailed assessment of plaque morphology and composition. However, it has limitations in accurately differentiating between different plaque components. On the other hand, OCT offers exceptional resolution, enabling precise characterization of plaque composition and features like fibrous cap thickness, and it also allows for the detection of thin-cap fibroatheromas, a high-risk plaque subtype[46]. However, the limited tissue penetration ability of OCT makes it less suitable for assessing deep plaque structures[47]. NIRS, on the contrary, provides information about the lipid content within plaques, aiding in identifying vulnerable lipid-rich plaques[48]. Nonetheless, NIRS may not have the same resolution level as IVUS or OCT[49]. In summary, while IVUS remains a standard tool, OCT and NIRS provide complementary insights into plaque regression analysis, enhancing our understanding of plaque composition and vulnerability.
Statin vs. placebo trials and observational cohorts
In 1997, the first IVUS randomized controlled trial (RCT) assessing the effect of statins on plaques demonstrated a significant reduction in TAV by −7% in the pravastatin group after 36 months, compared with a +41% increase in the placebo group (P < 0.01)[50]. Subsequently, one coronary computed tomographic angiography (CCTA) and four minor IVUS RCTs comparing statins to placebo obtained relatively similar results, but not all of them reached statistical significance[50]. In the largest observational study, a study to evaluate the effect of rosuvastatin on intravascular ultrasound-derived coronary atheroma burden (ASTEROID), 507 patients treated with a 40 mg dose of rosuvastatin showed a 6.1% reduction in TAV measured by IVUS at the end of 24 months[51]. Another large observational study, Coronary Atherosclerosis Study Measuring Effects of Rosuvastatin Using Intravascular Ultrasound in Japanese Subjects (COSMOS) in 2009, and the Integrated Biomarkers and Imaging Study (IBIS-4) in 2015, also demonstrated similar outcomes using IVUS[52].
The impact of diabetes mellitus on plaque progression was studied as part of the Phenotyping RElatives of persons with Diabetes with CGM and Tests of beta cell function and health (PREDICT) trial, in which plaque volume and characteristics under the influence of statins were compared between diabetic and non-diabetic patients, by assessing 61 patients with stable angina using IVUS[53]. The results indicated that the efficacy of statins for plaque regression might depend on comorbidity, as subjects with diabetes mellitus showed a significant increase in plaque volume and a higher presence of unstable plaque phenotypes.
Two large observational studies assessing the efficacy of statins, without differentiation between doses, did not show any signs of plaque regression but rather progression measured by coronary CT angiography[54]. Another observational study showed that among statin-treated patients with LDL-cholesterol levels less than 1.8 mmol/l, there was a decline in plaque progression (TAV +4.6%; P < 0.05) compared with patients with higher values (11.6%; P < 0.05), measured by coronary CT angiography[55].
High-intensity statin vs low-intensity statin trials
The efficacy of high-intensity statins for plaque regression has been established in all IVUS randomized trials comparing high-intensity statins to low-intensity statins. Three studies assessed major plaque regression among patients treated with high-intensity statins compared with control arms[56].
In 2004, The Reversal of Atherosclerosis with Aggressive Lipid Lowering trial (REVERSAL) was conducted to compare the treatment of atorvastatin and pravastatin using IVUS [57]. The study demonstrated that patients taking an 80 mg dose of atorvastatin had a reduction in TAV by −0.4% (P < 0.05), compared with +2.7% (P < 0.05) among patients treated with 40 mg of pravastatin at 18 months[57]. A similar comparison was observed between rosuvastatin 40 mg and atorvastatin 80 mg in the study of coronary atheroma by IVUS: the effect of rosuvastatin vs atorvastatin (SATURN), in which both treatment arms showed a reduction in TAV at 24 months, with −4.8% for rosuvastatin and −3.2% for atorvastatin (P < 0.05)[58]. However, the difference in primary outcome (changes in plaque area volume) was slight. In the Japan Assessment of Pitavastatin and Atorvastatin in Acute Coronary Syndrome (JAPAN-ACS) trial, atorvastatin demonstrated higher plaque regression results at the end of 12 months compared with pitavastatin; among 307 patients, those treated with a 20 mg dose of atorvastatin showed plaque regression by −18.1% (P < 0.05), while pitavastatin-treated patients at 4 mg showed regression by 16.9% (P < 0.05)[59].
Paradoxical results were reported by obtained in the Statin and Atheroma Vulnerability Evaluation (STABLE) trial, showing that a 10 mg dose of rosuvastatin was more effective for plaque regression than its higher dose (40 mg)[60]. Specifically, data measured by IVUS after 12 months of taking rosuvastatin 10 mg vs 40 mg showed reductions of −12.4% and −8.7%, respectively (P < 0.05). Furthermore, the multivariable analysis revealed that acute coronary syndrome, baseline TAV, and young age were correlated with a greater regression in TAV, and high values of necrotic core regression were correlated with baseline necrotic core volume, the presence of thin cap fibroatheroma, the baseline plaque burden, and the level of high-sensitivity C-reactive protein at follow-up[60].
Although plaque regression was observed in other studies, none of them found significant differences in the efficacy of high and low dosages[61].
Statin trials and plaque composition
Many studies have assessed the effects of statins on plaque composition, mostly using virtual histology IVUS or integrated backscatter IVUS. The majority of these studies showed that statin treatments increased calcified plaque and fibrous volume, but reduced non-calcified plaque volumes, fibrofatty volume, and necrotic core[62]. When virtual histology IVUS and OCT were used to evaluate fibrous cap thickness among statin-treated patients, the results showed a decreased thin-cap fibroatheromas but higher rates of fibroatheroma CT at follow-up [62]. The Prediction of Extent and Risk Profile of Coronary Atherosclerosis and Their Changes During Lipid-lowering Therapy Based on Non-invasive Techniques (PREDICT) study compared between diabetic and non-diabetic patients and found that the thin-cap fibroatheromas commonly remained present in subjects with diabetes[63].
NIRS and OCT have been used to evaluate lipid content. In the Reduction in Yellow Plaque by Aggressive Lipid-Lowering Therapy (YELLOW) trial, NIRS evaluation showed that the rosuvastatin (40mg) treated patients had much greater reductions in lipid content burden by −32.2% (P < 0.05) compared with −0.6% (P < 0.05) for patients treated with other statins[64]. The Effect of Early Pitavastatin Therapy on Coronary Fibrous Cap Thickness Assessed by OCT in Patients with Acute Coronary Syndrome (ESCORT) trial compared early and late pitavastatin treatment and showed a reduction in lipid content in both groups at 36 weeks measured by OCT; however, the fibroatheroma cap thickness increased by 20 μm in the early-treated group, compared with a decline of 5 μm in the late-treated group[65]. In the IBIS-4, the rosuvastatin (40 mg) treated patients showed reductions in the number of macrophages and a declined lipid content using OCT[66]. The results of the Effect of Atorvastatin Therapy on the Fibrous Cap Thickness in Coronary Atherosclerotic Plaque as Assessed by OCT (EASY-FIT) trial demonstrated that the higher the intensity of statin treatment, the greater increase in fibrous cap thickness and the greater decrease in lipid arc content, as assessed by OCT[67].
Recent studies using coronary CT angiography showed that statins had an effect on declining noncalcified plaque but elevating calcified plaque while evaluating plaque composition [68]. Moreover, the Progression of AtheRosclerotic PlAque Determined by Computed TomoGraphic Angiography Imaging (PARADIGM) trial demonstrated the effect of statins on declining high-risk plaque features, including positive arterial remodeling, spotty calcifications, or at least two features of low attenuation plaque[68].
The data obtained from numerous studies conclude that the effect of statins on plaque regression depends on their doses and is proportional to reductions in LDL cholesterol. IVUS has been the prevalent method for measuring plaque burden, but in recent years, coronary CT angiography has been used in some studies to evaluate changes[69]. Statins are effective in increasing calcified and fibrous plaque volumes but reducing fibrofatty, necrotic core, and non-calcified volumes[69].
Ezetimibe
The effectiveness of combining 10 mg ezetimibe with OMT for plaque regression was studied in several randomized trials using IVUS[70]. Most of these studies showed a significant decrease in plaque volume ranging from −2.9% to −13.9% in the ezetimibe group[70]. However, larger trials such as eZEtimibe Ultrasound Study (ZEUS), ezetimibe clinical investigation for regression of intracoronary plaque evaluated by angioscopy and ultrasound (ZIPANGU), Plaque Composition in Patients with acute ST Segment Elevation Myocardial Infarction assessed by Optical Coherence Tomography and IntraVascular UltraSound with iMap (OCTIVUS), Impact of Dual Lipid-Lowering Strategy With Ezetimibe and Atorvastatin on Coronary Plaque Regression in Patients With Percutaneous Coronary Intervention (PRECISE-IVUS), and Virtual histology evaluation of atherosclerosis regression during atorvastatin and ezetimibe administration (HEAVEN) did not demonstrate a significant difference between the treatment arms, despite a greater plaque regression observed in patients receiving ezetimibe in combination with OMT[71]. Additionally, Hibi et al found changes in plaque composition measured by IVUS, indicating a decline in fibrous and calcified plaques and an increase in lipid volume in the ezetimibe-treated group[72]. Although ezetimibe effectively lowers LDL cholesterol and reduces cardiovascular events, its efficacy for plaque regression remains uncertain[72]. Further research and larger-scale studies may be needed to establish its true effectiveness in this regard.
PCSK9 inhibitors
Recently, several RCTs have attempted to establish the effectiveness of PCSK9 inhibitors in plaque remodeling. PCSK9 plays a role in maintaining LDL homeostasis by reducing LDL receptors on liver cell membranes, which are responsible for removing LDL particles from the bloodstream[73]. Agents that block this enzyme decrease LDL particle concentrations in the blood[73]. The Global Assessment of Plaque Regression with PCSK9 Antibody as Measured by IVUS (GLAGOV) trial conducted in 2016, demonstrated a reduction in TAV of −2.9% (P < 0.05) in the evolocumab arm at 76 weeks, as assessed by IVUS; in comparison, the reduction was −0.4% (P < 0.05) in the OMT arm among the 968 randomized patients[74]. In 2019, the Evaluation of Effect of Alirocumab on Coronary Atheroma Volume in Japanese Patients Hospitalized for Acute Coronary Syndrome with Hypercholesterolaemia (ODYSSEY J-IVUS) trial randomized 206 patients with acute coronary syndrome into either the alirocumab 75 mg group or the OMT group; the results showed a lower TAV at 36 weeks in the experimental arm, but it did not reach statistical significance[75]. Further studies with extended follow-up periods and evaluations of plaque composition may provide more details on the unexpectedly low efficacy observed in the above-mentioned trials, compared with the effect on LDL cholesterol.
Cholesteryl ester transfer protein (CETP) inhibitors
The mechanism of action for CETP inhibitors in reducing LDL cholesterol is correlated with preventing the conversion of cholesterol from high-density lipoprotein (HDL) to LDL. However, the results obtained from several large clinical trials did not demonstrate significant clinical improvements[76]. In fact, some of these studies were terminated prematurely due to their lack of efficacy, despite observing positive changes in HDL and LDL levels;.nevertheless, the effect of CETP inhibitors on plaque volumes was evaluated in two studies[76]. In 2007, the randomized Investigation of Lipid Level Management Using Coronary Ultrasound to Assess Reduction of Atherosclerosis by CETP Inhibition and HDL Elevation (ILLUSTRATE) trial demonstrated a reduction in TAV of −4.8% (P < 0.05) in the torcetrapib group, compared with −3.2% (P < 0.05) in the OMT group, among 910 patients at the end of 24 months measured by IVUS; however, no significant results were obtained for plaque area volume[77]. Because of an increased morbidity and mortality from unspecified causes observed in a subsequent trial, further studies involving torcetrapib were discontinued. Another large trial showed no efficacy of dalcetrapib, although recent clinical trial results assessing anacetrapib appeared to be promising, but required further investigation regarding its effectiveness in plaque regression.
Omega-3 fatty acids
During the past few years, a modern and promising intervention strategy involving the use of omega-3 oils has been tested. This is because of the effects of eicosapentaenoic acid (EPA) and other omega-3 fatty acids in stabilizing endothelial dysfunction, which is closely correlated with the production of inflammatory mediators. Results obtained from several RCTs have demonstrated varying degrees of influence that EPA has on TAVs[78]. While one RCT using intravascular echocardiography and one using cardiac CT angiography did not provide clear evidence of EPA's effect on plaque regression, another RCT using integrated backscatter intravascular ultrasound (IB-IVUS) observed a greater regression of coronary plaques in the experimental arm receiving combination therapy of pitavastatin and EPA (1,800 mg/day) over a six-month period, compared with the control arm[79]. In these two intravascular echocardiography trials, patients receiving EPA treatment showed the decreased lipid volumes and improved fibrous volumes[78–79]. The results of an observational trial by Alfaddagh et al. in 2019 indicated that patients receiving eicosapentaenoic acid treatment with a serum level of at least 4% omega-3 oils experienced a decline in plaque progression as measured by cardiac CT angiography, compared with those with lower levels[80]. Numerous randomized and observational studies using OCT have also shown that EPA-treated patients exhibited an enhanced fibrous cap thickness[80]. Similarly, a study assessing neo-atherosclerotic lesions associated with percutaneous coronary intervention observed a similar effect of rosuvastatin and EPA on neoatherosclerosis[81]. In 2020, the Effect of Icosapent Ethyl on Progression of Coronary Atherosclerosis in Patients With Elevated Triglycerides on Statin Therapy (EVAPORATE) trial was conducted involving 80 randomized patients to evaluate the effect of a purified EPA ethyl ester or OMT on plaque regression, using cardiac CT angiography as a control[81]. At the end of the trial, a greater reduction in fibrous volume, fibrofatty volume, and low attenuation plaque was observed in the experimental arm, leading to significant plaque regression compared with the control arm (−9.0% vs. +11.0%; P < 0.05) at 18 months[81]. However, it is important to consider certain factors, such as the difference in plaque burden at baseline between both groups and the use of mineral oil by patients in the control group, which was associated with negative effects on apolipoprotein-B and LDL-C levels, as these factors may have influenced the trial results[81]. Overall, the initial results of treatment trials involving EPA are promising, and it is highly possible that EPA will become an integral part of plaque regression strategies in clinical practice in the future.
Lipoprotein(a) and agents lowering triglycerides
Despite some observational studies suggesting a connection between levels of lipoprotein(a) [Lp(a)] and high-risk plaque features and plaque volume, there are limited data about the effect of Lp(a)-lowering agents on plaque composition and volume. A small RCT using OCT did not find any difference in the percentage of atheroma volume in saphenous vein grafts when evaluating the effect of niacin, which may lower Lp(a) by approximately 25%[82]. Because of its limited efficacy and significant side effects, niacin is not commonly used in clinical practice[82]. Another approach to reduce Lp(a) by approximately 25% is through the use of PCSK9 inhibitors, which may have positive effects on reducing plaque volumes[83]. Currently, there are no available data on the efficacy of triglyceride-lowering agents such as fenofibrates, although a RCT using IVUS is currently underway[83].
Antihypertensive drugs
Among several studies evaluating the influence of antihypertensive drugs on plaque volumes using IVUS, only one of them demonstrated some evidence of plaque regression. In the Comparison of Amlodipine vs Enalapril to Limit Occurrences of Thrombosis study conducted in 2004, 274 patients were randomized into enalapril, amlodipine, and placebo groups[84], in which the evaluation of percent atheroma volumes at 24 months showed a similar effect in each study group, with a partial decline in plaque progression in the amlodipine and enalapril arms, compared with the control arm (+0.5% for amlodipine vs. +1.3% for placebo, P < 0.05).
In the Impact of Olmesarten on Progression of Coronary Atherosclerosis: Evaluation by IVUS trial conducted in 2010, 247 patients were randomized into olmesartan or placebo groups[85]. At the end of 14 months, the IVUS results demonstrated a reduction in plaque progression in the olmesartan group (−0.6% compared with +5.4% in the control arm, P < 0.05)[85]. However, three other randomized trials (Perindopril's Prospective Effect on Coronary Atherosclerosis by Angiography and IVUS Evaluation trial in 2007[86], Han et al. trial in 2012[87], and the Aliskiren Quantitative Atherosclerosis Regression IVUS Study in 2013[88]) did not find any significant effect of antihypertensive drugs (perindopril, ramipril, and aliskiren, respectively) on plaque progression compared with control arms.
Colchicine
Colchicine, a medicine with anti-inflammatory effects, inhibits microtubule polymerization by binding to tubulin, thereby suppressing the inflammatory response. In a study conducted in 2017, 80 patients with a recent history of acute coronary syndrome were treated with OMT or a daily dose of 0.5 mg of colchicine in an open-label, nonrandomized study that utilized coronary CT angiography[89]. The results obtained from this study at the end of 12 months demonstrated similar reductions in TAV in both treatment groups (−17.7% vs. −10.8%; P > 0.05); however, it was observed that colchicine treatment led to a significant decline in non-calcified plaque volumes and low attenuation plaque volumes (15.9 mm3vs. 6.6 mm3; P < 0.05)[89]. Since the efficacy of colchicine on coronary artery disease treatment has been observed in recent[90], there is an undeniable need for further trials that evaluate the influence of colchicine on the composition and regression of plaques.
Anticoagulants
Several studies were conducted to determine the effect of warfarin and direct oral anticoagulants on plaque progression. In 2019, Lee et al[91] conducted a study with 97 patients experiencing atrial fibrillation, and the participants were randomized into rivaroxaban or warfarin groups, and the results obtained from cardiac CT angiography after 12 months demonstrated an increase in the total plaque volume caused by warfarin[90]. In 2019, Beyer et al[92] conducted an observational cohort study with 161 participants using cardiac CT angiography and detected a decrease in absolute plaque burden by −7.1 mm3 (P < 0.05) after 38 months among patients who were taking direct acting oral anticoagulants. Patients receiving warfarin treatment and control subjects demonstrated changes in absolute plaque burden of +66.5 mm3 and +27.2 mm3, respectively (P < 0.05). In contrast, a post hoc analysis of eight randomized trials by Andrews et al[93] in 2018 suggested that warfarin treatment increased coronary calcification without affecting plaque volume.
It is difficult to draw firm conclusions about the efficacy of anticoagulants based on these data, as they are either observational or collected from small cohorts.
Oral Hypoglycemic Agents (OHAs)
Some data suggest a connection between plaque regression and thiazolidinedione treatment in patients with diabetes. The studies conducted by Yang et al. and the PERISCOPE RCTs obtained similar results, demonstrating the impact of pioglitazone on plaque regression[94]. Patients receiving pioglitazone treatment showed greater rates of decreasing thin-cap fibroatheroma and plaque regression, as measured by IVUS, compared with the control group after 6 and 18 months (PERISCOPE)[94]. Similarly, the APPROACH IVUS RCT found that patients receiving rosiglitazone demonstrated a reduction in TAV, compared with patients treated with glipizide, with no significant difference in percent atheroma volume[95]. However, it is important to consider the late data regarding the cardiovascular risks associated with rosiglitazone and the decline in its widespread usage. Given that recent trials of sodium-glucose cotransporter 2 (SGLT2) inhibitors have shown beneficial effects on patients with coronary artery disease and heart failure, future studies examining their influence on plaques and plaque regression are highly anticipated[96].
Other treatments
IVUS has been used to study the efficacy of various treatments on plaque burden. In the Low-Density Lipoprotein-Apheresis Coronary Morphology and Reserve Trial (LACMART) in 2002, 18 patients were randomized to receive either LDL-apheresis treatment or usual care[97]. At the end of 12 months, the treatment group demonstrated plaque regression of −8.2%, compared with +12.4% in the control group (P < 0.05). However, because of the complexity of LDL-apheresis and the availability of easier-to-use methods such as statins, it is unlikely for this method to become widely prevalent in clinical practice.
In the ApoA-1 Milano trial, apolipoprotein A1-based HDL mimetics demonstrated plaque regression of −5.0%, compared with +1.7% in the control group (P < 0.05)[98]. However, subsequent findings from a larger CHI-SQUARE trial using IVUS disproved those results.
Several treatments, including the CB1 antagonist rimonabant, acyl-CoA:cholesterol acyltransferase (ACAT) inhibitor pactimibe, HDL delipidation, reconstituted HDL infusions, probucol, succinobucol, and a Bromodomain and Extra-Terminal motif (BET) protein inhibitor, were found to be ineffective for plaque regression in the Strategy to Reduce Atherosclerosis Development Involving Administration of Rimonabant—The Intravascular Ultrasound Study (STRADIVARIUS)[98], ACAT Intravascular Atherosclerosis Treatment Evaluation (ACTIVATE)[99], reconstituted HDL infusions[100], Synergistic Effect of Combination Therapy with Cilostazol and ProbUcol on Plaque Stabilization and Lesion REgression (SECURE)[101], Canadian Antioxidant Restenosis Trial 2 (CART-2), and ApoA-I Synthesis Stimulation and Intravascular Ultrasound for Coronary Atheroma Regression Evaluation (ASSURE)[102] IVUS randomized trials, respectively.
A RCT assessing the correlation between testosterone and plaque progression among subjects with hypogonadism showed a greater coronary plaque volume progression in patients receiving testosterone treatment[103]. In two smaller RCTs, the aged garlic extract demonstrated low attenuation plaque progression and coronary artery calcification (CAC) reduction in the treatment groups[104].
Conclusions and perspectives
In this review, we have summarized the main methods and techniques that reverse the progression of atherosclerosis and lead to plaque regression in the coronary arteries. Visualization of atherosclerotic plaques plays a significant role in the treatment. The effectiveness of the treatment is associated with the timely initiation of therapy, which is often hindered by the absence of symptoms and challenges in diagnosing atherosclerotic lesions. Based on the available data, it can be concluded that statins are currently the most effective means of combating atherosclerosis and promoting plaque regression. However, statins are not a flawless treatment for atherosclerosis, and despite their widespread use and recommendations, future investigations are needed to identify more effective strategies.
Acknowledgments
None.
Fundings
This research was funded by Russian Science Foundation, grant number 23-25-00339.
Dave T, Ezhilan J, Vasnawala H, et al. Plaque regression and plaque stabilisation in cardiovascular diseases[J]. Indian J Endocrinol Metab, 2013, 17(6): 983–989. doi: 10.4103/2230-8210.122604
[5]
Feig JE. Regression of atherosclerosis: insights from animal and clinical studies[J]. Ann Glob Health, 2014, 80(1): 13–23. doi: 10.1016/j.aogh.2013.12.001
[6]
Katra P, Björkbacka H. Atherosclerosis: cell biology and lipoproteins[J]. Curr Opin Lipidol, 2022, 33(3): 208–210. doi: 10.1097/MOL.0000000000000815
[7]
Borén J, Chapman MJ, Krauss RM, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel[J]. Eur Heart J, 2020, 41(24): 2313–2330. doi: 10.1093/eurheartj/ehz962
[8]
Gerhardt T, Haghikia A, Stapmanns P, et al. Immune mechanisms of plaque instability[J]. Front Cardiovasc Med, 2022, 8: 797046. doi: 10.3389/fcvm.2021.797046
[9]
Li Y, Deng S, Liu B, et al. The effects of lipid-lowering therapy on coronary plaque regression: a systematic review and meta-analysis[J]. Sci Rep, 2021, 11(1): 7999. doi: 10.1038/s41598-021-87528-w
[10]
Rocha VZ, Rached FH, Miname MH. Insights into the role of inflammation in the management of atherosclerosis[J]. J Inflamm Res, 2023, 16: 2223–2239. doi: 10.2147/JIR.S276982
[11]
Silva GM, França-Falcão MS, Calzerra NTM, et al. Role of renin-angiotensin system components in atherosclerosis: focus on Ang-II, ACE2, and Ang-1–7[J]. Front Physiol, 2020, 11: 1067. doi: 10.3389/fphys.2020.01067
[12]
Waring OJ, Skenteris NT, Biessen EAL, et al. Two-faced Janus: the dual role of macrophages in atherosclerotic calcification[J]. Cardiovasc Res, 2022, 118(13): 2768–2777. doi: 10.1093/cvr/cvab301
[13]
Kowara M, Cudnoch-Jedrzejewska A. Different approaches in therapy aiming to stabilize an unstable atherosclerotic plaque[J]. Int J Mol Sci, 2021, 22(9): 4354. doi: 10.3390/ijms22094354
[14]
Li Y, Luo X, Hua Z, et al. Apolipoproteins as potential communicators play an essential role in the pathogenesis and treatment of early atherosclerosis[J]. Int J Biol Sci, 2023, 19(14): 4493–4510. doi: 10.7150/ijbs.86475
[15]
Poznyak AV, Silaeva YY, Orekhov AN, et al. Animal models of human atherosclerosis: current progress[J]. Braz J Med Biol Res, 2020, 53(6): e9557. doi: 10.1590/1414-431x20209557
[16]
Harman JL, Jørgensen HF. The role of smooth muscle cells in plaque stability: Therapeutic targeting potential[J]. Br J Pharmacol, 2019, 176(19): 3741–3753. doi: 10.1111/bph.14779
[17]
Phu TA, Ng M, Vu NK, et al. ApoE expression in macrophages communicates immunometabolic signaling that controls hyperlipidemia-driven hematopoiesis & inflammation via extracellular vesicles[J]. J Extracell Vesicles, 2023, 12(8): e12345. doi: 10.1002/jev2.12345
[18]
Zhao R, Liu H, Zhang S, et al. A novel animal model for vulnerable atherosclerotic plaque: dehydrated ethanol lavage in the carotid artery of rabbits fed a Western diet[J]. Cardiovasc Diagn Ther, 2021, 11(6): 1241–1252. doi: 10.21037/cdt-21-291
[19]
Patel S, Mastrogiacomo L, Fulmer M, et al. Deletion of macrophage-specific glycogen synthase kinase (GSK)-3α promotes atherosclerotic regression in Ldlr−/− mice[J]. Int J Mol Sci, 2022, 23(16): 9293. doi: 10.3390/ijms23169293
[20]
Yanai H, Adachi H, Hakoshima M, et al. Atherogenic lipoproteins for the statin residual cardiovascular disease risk[J]. Int J Mol Sci, 2022, 23(21): 13499. doi: 10.3390/ijms232113499
[21]
Rong JX, Li J, Reis ED, et al. Elevating high-density lipoprotein cholesterol in apolipoprotein E-deficient mice remodels advanced atherosclerotic lesions by decreasing macrophage and increasing smooth muscle cell content[J]. Circulation, 2001, 104(20): 2447–2452. doi: 10.1161/hc4501.098952
[22]
Feig JE, Parathath S, Rong JX, et al. Reversal of hyperlipidemia with a genetic switch favorably affects the content and inflammatory state of macrophages in atherosclerotic plaques[J]. Circulation, 2011, 123(9): 989–998. doi: 10.1161/CIRCULATIONAHA.110.984146
[23]
Kawashiri MA, Zhang Y, Usher D, et al. Effects of coexpression of the LDL receptor and apoE on cholesterol metabolism and atherosclerosis in LDL receptor-deficient mice[J]. J Lipid Res, 2001, 42(6): 943–950. doi: 10.1016/S0022-2275(20)31618-7
[24]
Cho KH, Hong YJ. Proprotein convertase subtilisin/kexin type 9 inhibition in cardiovascular disease: current status and future perspectives[J]. Korean J Intern Med, 2020, 35(5): 1045–1058. doi: 10.3904/kjim.2020.140
[25]
Barrett TJ. Macrophages in atherosclerosis regression[J]. Arterioscler Thromb Vasc Biol, 2020, 40(1): 20–33. doi: 10.1161/ATVBAHA.119.312802
[26]
Judd J, Lovas J, Huang GN. Defined factors to reactivate cell cycle activity in adult mouse cardiomyocytes[J]. Sci Rep, 2019, 9(1): 18830. doi: 10.1038/s41598-019-55027-8
[27]
Simo OK, Berrougui H, Fulop T, et al. The susceptibility to diet-induced atherosclerosis is exacerbated with aging in C57B1/6 mice[J]. Biomedicines, 2021, 9(5): 487. doi: 10.3390/biomedicines9050487
[28]
Dominguez LJ, Veronese N, Vernuccio L, et al. Nutrition, physical activity, and other lifestyle factors in the prevention of cognitive decline and dementia[J]. Nutrients, 2021, 13(11): 4080. doi: 10.3390/nu13114080
[29]
Meiring RM, Tanimukai K, Bradnam L. The effect of exercise-based cardiac rehabilitation on objectively measured physical activity and sedentary behavior: a systematic review and meta-analysis[J]. J Prim Care Community Health, 2020, 11: 2150132720935290.
[30]
Geng L, Yuan Y, Du P, et al. The association between intravascular ultrasound-derived echo-attenuation and quantitative flow ratio in intermediate coronary lesions[J]. Cardiovasc Diagn Ther, 2021, 11(6): 1206–1216. doi: 10.21037/cdt-21-402
[31]
De Bosscher R, Dausin C, Claus P, et al. Endurance exercise and the risk of cardiovascular pathology in men: a comparison between lifelong and late-onset endurance training and a non-athletic lifestyle - rationale and design of the Master@Heart study, a prospective cohort trial[J]. BMJ Open Sport Exerc Med, 2021, 7(2): e001048. doi: 10.1136/bmjsem-2021-001048
[32]
Yang S, Zeng Z, Yuan Q, et al. Vascular calcification: from the perspective of crosstalk[J]. Mol Biomed, 2023, 4(1): 35. doi: 10.1186/s43556-023-00146-y
[33]
Ngamdu KS, Ghosalkar DS, Chung HE, et al. Long-term statin therapy is associated with severe coronary artery calcification[J]. PLoS One, 2023, 18(7): e0289111. doi: 10.1371/journal.pone.0289111
[34]
Pulipati VP, Alenghat FJ. The impact of lipid-lowering medications on coronary artery plaque characteristics[J]. Am J Prev Cardiol, 2021, 8: 100294. doi: 10.1016/j.ajpc.2021.100294
[35]
Xian JZ, Lu M, Fong F, et al. Statin effects on vascular calcification: microarchitectural changes in aortic calcium deposits in aged hyperlipidemic mice[J]. Arterioscler Thromb Vasc Biol, 2021, 41(4): e185–e192. doi: 10.1161/ATVBAHA.120.315737
[36]
Sallam T, Tintut Y, Demer LL. Regulation of calcific vascular and valvular disease by nuclear receptors[J]. Curr Opin Lipidol, 2019, 30(5): 357–363. doi: 10.1097/MOL.0000000000000632
[37]
Henzel J, Kępka C, Kruk M, et al. High-risk coronary plaque regression after intensive lifestyle intervention in nonobstructive coronary disease: a randomized study[J]. JACC Cardiovasc Imaging, 2021, 14(6): 1192–1202. doi: 10.1016/j.jcmg.2020.10.019
[38]
Ornish D, Scherwitz LW, Billings JH, et al. Intensive lifestyle changes for reversal of coronary heart disease[J]. JAMA, 1998, 280(23): 2001–2007. doi: 10.1001/jama.280.23.2001
[39]
Wu AD, Lindson N, Hartmann-Boyce J, et al. Smoking cessation for secondary prevention of cardiovascular disease[J]. Cochrane Database Syst Rev, 2022, 8(8): CD014936. https://pubmed.ncbi.nlm.nih.gov/35938889/
[40]
Laguzzi F, Baldassarre D, Veglia F, et al. Alcohol consumption in relation to carotid subclinical atherosclerosis and its progression: results from a European longitudinal multicentre study[J]. Eur J Nutr, 2021, 60(1): 123–134. doi: 10.1007/s00394-020-02220-5
[41]
Hata Y, Mochizuki J, Okamoto S, et al. Aortic calcification is associated with coronary artery calcification and is a potential surrogate marker for ischemic heart disease risk: A cross-sectional study[J]. Medicine (Baltimore), 2022, 101(29): e29875. doi: 10.1097/MD.0000000000029875
[42]
US Preventive Services Task Force, Mangione CM, Barry MJ, et al. Statin use for the primary prevention of cardiovascular disease in adults: US preventive services task force recommendation statement[J]. JAMA, 2022, 328(8): 746–753. doi: 10.1001/jama.2022.13044
[43]
Toth PP, Banach M. Statins: then and now[J]. Methodist Debakey Cardiovasc J, 2019, 15(1): 23–31. doi: 10.14797/mdcj-15-1-23
[44]
Wakabayashi K, Nozue T, Yamamoto S, et al. Efficacy of statin therapy in inducing coronary plaque regression in patients with low baseline cholesterol levels[J]. J Atheroscler Thromb, 2016, 23(9): 1055–1066. doi: 10.5551/jat.34660
[45]
Legutko J, Bryniarski KL, Kaluza GL, et al. Intracoronary imaging of vulnerable plaque-from clinical research to everyday practice[J]. J Clin Med, 2022, 11(22): 6639. doi: 10.3390/jcm11226639
[46]
Daghem M, Bing R, Fayad ZA, et al. Noninvasive imaging to assess atherosclerotic plaque composition and disease activity: coronary and carotid applications[J]. JACC Cardiovasc Imaging, 2020, 13(4): 1055–1068. doi: 10.1016/j.jcmg.2019.03.033
[47]
Araki M, Park SJ, Dauerman HL, et al. Optical coherence tomography in coronary atherosclerosis assessment and intervention[J]. Nat Rev Cardiol, 2022, 19(10): 684–703. doi: 10.1038/s41569-022-00687-9
[48]
Kitahara S, Kataoka Y, Sugane H, et al. In vivo imaging of vulnerable plaque with intravascular modalities: its advantages and limitations[J]. Cardiovasc Diagn Ther, 2020, 10(5): 1461–1479. doi: 10.21037/cdt-20-238
[49]
Nagaraja V, Kalra A, Puri R. When to use intravascular ultrasound or optical coherence tomography during percutaneous coronary intervention?[J]. Cardiovasc Diagn Ther, 2020, 10(5): 1429–1444. doi: 10.21037/cdt-20-206
[50]
Takagi T, Yoshida K, Akasaka T, et al. Intravascular ultrasound analysis of reduction in progression of coronary narrowing by treatment with pravastatin[J]. Am J Cardiol, 1997, 79(12): 1673–1676. doi: 10.1016/S0002-9149(97)00221-X
[51]
Chhatriwalla AK, Nicholls SJ, Nissen SE. The ASTEROID trial: coronary plaque regression with high-dose statin therapy[J]. Future Cardiol, 2006, 2(6): 651–654. doi: 10.2217/14796678.2.6.651
[52]
Adachi T, Ohsuzu F. Cosmic effect of rosuvastatin in COSMOS[J]. Circ J, 2009, 73(11): 2015–2016. doi: 10.1253/circj.CJ-09-0700
[53]
Kovarnik T, Chen Z, Mintz GS, et al. Plaque volume and plaque risk profile in diabetic vs. non-diabetic patients undergoing lipid-lowering therapy: a study based on 3D intravascular ultrasound and virtual histology[J]. Cardiovasc Diabetol, 2017, 16(1): 156. doi: 10.1186/s12933-017-0637-0
[54]
Kazemian P, Wexler DJ, Fields NF, et al. Development and validation of PREDICT-DM: a new microsimulation model to project and evaluate complications and treatments of type 2 diabetes mellitus[J]. Diabetes Technol Ther, 2019, 21(6): 344–355. doi: 10.1089/dia.2018.0393
[55]
Gao D, Hua R, Jiesisibieke D, et al. C-reactive protein and coronary atheroma regression following statin therapy: A meta-regression of randomized controlled trials[J]. Front Cardiovasc Med, 2022, 9: 989527. doi: 10.3389/fcvm.2022.989527
[56]
Nissen SE, Nicholls SJ, Sipahi I, et al. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial[J]. JAMA, 2006, 295(13): 1556–1565. doi: 10.1001/jama.295.13.jpc60002
[57]
Nissen SE. Effect of intensive lipid lowering on progression of coronary atherosclerosis: evidence for an early benefit from the Reversal of Atherosclerosis with Aggressive Lipid Lowering (REVERSAL) trial[J]. Am J Cardiol, 2005, 96(S5): 61–68. https://www.sciencedirect.com/science/article/abs/pii/S0002914905011379
[58]
Puri R, Ballantyne CM, Hoogeveen RC, et al. Lipoprotein(a) and coronary atheroma progression rates during long-term high-intensity statin therapy: Insights from SATURN[J]. Atherosclerosis, 2017, 263: 137–144. doi: 10.1016/j.atherosclerosis.2017.06.026
[59]
Hibi K, Kimura T, Kimura K, et al. Clinically evident polyvascular disease and regression of coronary atherosclerosis after intensive statin therapy in patients with acute coronary syndrome: serial intravascular ultrasound from the Japanese assessment of pitavastatin and atorvastatin in acute coronary syndrome (JAPAN-ACS) trial[J]. Atherosclerosis, 2011, 219(2): 743–749. doi: 10.1016/j.atherosclerosis.2011.08.024
[60]
Park SJ, Kang SJ, Ahn JM, et al. Effect of statin treatment on modifying plaque composition: a double-blind, randomized study[J]. J Am Coll Cardiol, 2016, 67(15): 1772–1783. doi: 10.1016/j.jacc.2016.02.014
[61]
Gaba P, Gersh BJ, Muller J, et al. Evolving concepts of the vulnerable atherosclerotic plaque and the vulnerable patient: implications for patient care and future research[J]. Nat Rev Cardiol, 2023, 20(3): 181–196. doi: 10.1038/s41569-022-00769-8
[62]
Gu SZ, Costopoulos C, Huang Y, et al. High-intensity statin treatment is associated with reduced plaque structural stress and remodelling of artery geometry and plaque architecture[J]. Eur Heart J Open, 2021, 1(3): oeab039. doi: 10.1093/ehjopen/oeab039
[63]
Dawson LP, Lum M, Nerleker N, et al. Coronary atherosclerotic plaque regression: JACC state-of-the-art review[J]. J Am Coll Cardiol, 2022, 79(1): 66–82. doi: 10.1016/j.jacc.2021.10.035
[64]
Kini AS, Baber U, Kovacic JC, et al. Changes in plaque lipid content after short-term intensive versus standard statin therapy: the YELLOW trial (reduction in yellow plaque by aggressive lipid-lowering therapy)[J]. J Am Coll Cardiol, 2013, 62(1): 21–29. doi: 10.1016/j.jacc.2013.03.058
[65]
Luo H, Lu J, Bai Y, et al. Effect of camrelizumab vs placebo added to chemotherapy on survival and progression-free survival in patients with advanced or metastatic esophageal squamous cell carcinoma: the ESCORT-1st randomized clinical trial[J]. JAMA, 2021, 326(10): 916–925. doi: 10.1001/jama.2021.12836
[66]
Taniwaki M, Radu MD, Garcia-Garcia HM, et al. Long-term safety and feasibility of three-vessel multimodality intravascular imaging in patients with ST-elevation myocardial infarction: the IBIS-4 (integrated biomarker and imaging study) substudy[J]. Int J Cardiovasc Imaging, 2015, 31(5): 915–926. doi: 10.1007/s10554-015-0631-0
[67]
Komukai K, Kubo T, Kitabata H, et al. Effect of atorvastatin therapy on fibrous cap thickness in coronary atherosclerotic plaque as assessed by optical coherence tomography: the EASY-FIT study[J]. J Am Coll Cardiol, 2014, 64(21): 2207–2217. doi: 10.1016/j.jacc.2014.08.045
[68]
Won KB, Lee SE, Lee BK, et al. Longitudinal assessment of coronary plaque volume change related to glycemic status using serial coronary computed tomography angiography: A PARADIGM (Progression of AtheRosclerotic PlAque DetermIned by Computed TomoGraphic Angiography Imaging) substudy[J]. J Cardiovasc Comput Tomogr, 2019, 13(2): 142–147. doi: 10.1016/j.jcct.2018.12.002
[69]
Nakazato R, Gransar H, Berman DS, et al. Statins use and coronary artery plaque composition: results from the International Multicenter CONFIRM Registry[J]. Atherosclerosis, 2012, 225(1): 148–153. doi: 10.1016/j.atherosclerosis.2012.08.002
[70]
Reddy V, Allison J, Mounsey A. Is there benefit to adding ezetimibe to a statin for the secondary prevention of CVD?[J]. J Fam Pract, 2023, 72(5): 227–229. doi: 10.12788/jfp.0610
[71]
Jin J, Shan L, Wang M, et al. Variability in plasma lipids between intensive statin therapy and conventional-dose statins combined with ezetimibe therapy in patients with coronary atherosclerosis disease[J]. Int Heart J, 2023, 64(5): 807–815. doi: 10.1536/ihj.23-125
[72]
Hibi K, Sonoda S, Kawasaki M, et al. Effects of ezetimibe-statin combination therapy on coronary atherosclerosis in acute coronary syndrome[J]. Circ J, 2018, 82(3): 757–766. doi: 10.1253/circj.CJ-17-0598
[73]
Maligłówka M, Kosowski M, Hachuła M, et al. Insight into the evolving role of PCSK9[J]. Metabolites, 2022, 12(3): 256. doi: 10.3390/metabo12030256
[74]
Puri R, Nissen SE, Somaratne R, et al. Impact of PCSK9 inhibition on coronary atheroma progression: Rationale and design of Global Assessment of Plaque Regression with a PCSK9 Antibody as Measured by Intravascular Ultrasound (GLAGOV)[J]. Am Heart J, 2016, 176: 83–92. doi: 10.1016/j.ahj.2016.01.019
[75]
Ako J, Hibi K, Tsujita K, et al. Effect of alirocumab on coronary atheroma volume in japanese patients with acute coronary syndrome- the ODYSSEY J-IVUS trial[J]. Circ J, 2019, 83(10): 2025–2033. doi: 10.1253/circj.CJ-19-0412
[76]
Nurmohamed NS, Ditmarsch M, Kastelein JJP. Cholesteryl ester transfer protein inhibitors: from high-density lipoprotein cholesterol to low-density lipoprotein cholesterol lowering agents?[J]. Cardiovasc Res, 2022, 118(14): 2919–2931. doi: 10.1093/cvr/cvab350
[77]
Su X, Li G, Deng Y, et al. Cholesteryl ester transfer protein inhibitors in precision medicine[J]. Clin Chim Acta, 2020, 510: 733–740. doi: 10.1016/j.cca.2020.09.012
[78]
Sherratt SCR, Libby P, Budoff MJ, et al. Role of omega-3 fatty acids in cardiovascular disease: the debate continues[J]. Curr Atheroscler Rep, 2023, 25(1): 1–17. doi: 10.1007/s11883-022-01075-x
[79]
Baruś P, Modrzewski J, Gumiężna K, et al. Comparative appraisal of intravascular ultrasound and optical coherence tomography in invasive coronary imaging: 2022 update[J]. J Clin Med, 2022, 11(14): 4055. doi: 10.3390/jcm11144055
[80]
Alfaddagh A, Elajami TK, Saleh M, et al. An omega-3 fatty acid plasma index ≥4% prevents progression of coronary artery plaque in patients with coronary artery disease on statin treatment[J]. Atherosclerosis, 2019, 285: 153–162. doi: 10.1016/j.atherosclerosis.2019.04.213
[81]
Budoff MJ, Bhatt DL, Kinninger A, et al. Effect of icosapent ethyl on progression of coronary atherosclerosis in patients with elevated triglycerides on statin therapy: final results of the EVAPORATE trial[J]. Eur Heart J, 2020, 41(40): 3925–3932. doi: 10.1093/eurheartj/ehaa652
[82]
Kaiser Y, Daghem M, Tzolos E, et al. Association of lipoprotein(a) with atherosclerotic plaque progression[J]. J Am Coll Cardiol, 2022, 79(3): 223–233. doi: 10.1016/j.jacc.2021.10.044
[83]
Zivkovic S, Maric G, Cvetinovic N, et al. Anti-inflammatory effects of lipid-lowering drugs and supplements-a narrative review[J]. Nutrients, 2023, 15(6): 1517. doi: 10.3390/nu15061517
[84]
Nissen SE, Tuzcu EM, Libby P, et al. Effect of antihypertensive agents on cardiovascular events in patients with coronary disease and normal blood pressure: the CAMELOT study: a randomized controlled trial[J]. JAMA, 2004, 292(18): 2217–2225. doi: 10.1001/jama.292.18.2217
[85]
Hirohata A, Yamamoto K, Miyoshi T, et al. Impact of olmesartan on progression of coronary atherosclerosis: a serial volumetric intravascular ultrasound analysis from the OLIVUS (impact of OLmesarten on progression of coronary atherosclerosis: evaluation by intravascular ultrasound) trial[J]. J Am Coll Cardiol, 2010, 55(10): 976–982. doi: 10.1016/j.jacc.2009.09.062
[86]
Rodriguez-Granillo GA, Vos J, Bruining N, et al. Long-term effect of perindopril on coronary atherosclerosis progression (from the perindopril's prospective effect on coronary atherosclerosis by angiography and intravascular ultrasound evaluation study)[J]. Am J Cardiol, 2007, 100(2): 159–163. doi: 10.1016/j.amjcard.2007.02.073
[87]
Han C, Wang Q, Meng PP, et al. Effects of intensity of arm training on hemiplegic upper extremity motor recovery in stroke patients: a randomized controlled trial[J]. Clin Rehabil, 2013, 27(1): 75–81. doi: 10.1177/0269215512447223
[88]
Nicholls SJ, Bakris GL, Kastelein JJP, et al. Effect of aliskiren on progression of coronary disease in patients with prehypertension: the AQUARIUS randomized clinical trial[J]. JAMA, 2013, 310(11): 1135–1144. doi: 10.1001/jama.2013.277169
[89]
Zhang FS, He QZ, Qin CH, et al. Therapeutic potential of colchicine in cardiovascular medicine: a pharmacological review[J]. Acta Pharmacol Sin, 2022, 43(9): 2173–2190. doi: 10.1038/s41401-021-00835-w
[90]
Tong DC, Quinn S, Nasis A, et al. Colchicine in patients with acute coronary syndrome: the Australian COPS randomized clinical trial[J]. Circulation, 2020, 142(20): 1890–1900. doi: 10.1161/CIRCULATIONAHA.120.050771
[91]
Lee SR, Choi EK, Park SH, et al. Comparing warfarin and 4 direct oral anticoagulants for the risk of dementia in patients with atrial fibrillation[J]. Stroke, 2021, 52(11): 3459–3468. doi: 10.1161/STROKEAHA.120.033338
[92]
Beyer C, Tokarska L, Stühlinger M, et al. Structural cardiac remodeling in atrial fibrillation[J]. JACC Cardiovasc Imaging, 2021, 14(11): 2199–2208. doi: 10.1016/j.jcmg.2021.04.027
[93]
Andrews RK, Gardiner EE. Monitoring the pulse of thrombus formation: Comment on "Modeling thrombosis in silico: Frontiers, challenges, unresolved problems and milestones" by A. V. Belyaev et al[J]. Phys Life Rev, 2018, 26–27: 113–115.
[94]
Nicholls SJ, Tuzcu EM, Wolski K, et al. Lowering the triglyceride/high-density lipoprotein cholesterol ratio is associated with the beneficial impact of pioglitazone on progression of coronary atherosclerosis in diabetic patients: insights from the PERISCOPE (Pioglitazone Effect on Regression of Intravascular Sonographic Coronary Obstruction Prospective Evaluation) study[J]. J Am Coll Cardiol, 2011, 57(2): 153–159. doi: 10.1016/j.jacc.2010.06.055
[95]
Zeliadt SB, Coggeshall S, Thomas E, et al. The APPROACH trial: Assessing pain, patient-reported outcomes, and complementary and integrative health[J]. Clin Trials, 2020, 17(4): 351–359. doi: 10.1177/1740774520928399
[96]
Chambergo-Michilot D, Tauma-Arrué A, Loli-Guevara S. Effects and safety of SGLT2 inhibitors compared to placebo in patients with heart failure: A systematic review and meta-analysis[J]. Int J Cardiol Heart Vasc, 2020, 32: 100690. https://pubmed.ncbi.nlm.nih.gov/33335975/
[97]
Matsuzaki M, Hiramori K, Imaizumi T, et al. Intravascular ultrasound evaluation of coronary plaque regression by low density lipoprotein-apheresis in familial hypercholesterolemia: the Low Density Lipoprotein-Apheresis Coronary Morphology and Reserve Trial (LACMART)[J]. J Am Coll Cardiol, 2002, 40(2): 220–227. doi: 10.1016/S0735-1097(02)01955-1
[98]
Nissen SE, Tsunoda T, Tuzcu EM, et al. Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial[J]. JAMA, 2003, 290(17): 2292–2300. doi: 10.1001/jama.290.17.2292
[99]
Nissen SE, Nicholls SJ, Wolski K, et al. Effect of rimonabant on progression of atherosclerosis in patients with abdominal obesity and coronary artery disease: the STRADIVARIUS randomized controlled trial[J]. JAMA, 2008, 299(13): 1547–1560. doi: 10.1001/jama.299.13.1547
[100]
Giamarellos-Bourboulis EJ, Tsilika M, Moorlag S, et al. Activate: randomized clinical trial of BCG vaccination against infection in the elderly[J]. Cell, 2020, 183(2): 315–323.e9. doi: 10.1016/j.cell.2020.08.051
[101]
Tardiff BE, Newman MF, Saunders AM, et al. Preliminary report of a genetic basis for cognitive decline after cardiac operations. The Neurologic Outcome Research Group of the Duke Heart Center[J]. Ann Thorac Surg, 1997, 64(3): 715–720. doi: 10.1016/S0003-4975(97)00757-1
[102]
Castellano JM, Pocock SJ, Bhatt DL, et al. Polypill strategy in secondary cardiovascular prevention[J]. N Engl J Med, 2022, 387(11): 967–977. doi: 10.1056/NEJMoa2208275
[103]
Haas NB, Manola J, Dutcher JP, et al. Adjuvant treatment for high-risk clear cell renal cancer: updated results of a high-risk subset of the ASSURE randomized trial[J]. JAMA Oncol, 2017, 3(9): 1249–1252. doi: 10.1001/jamaoncol.2017.0076
[104]
Budoff MJ, Ellenberg SS, Lewis CE, et al. Testosterone treatment and coronary artery plaque volume in older men with low testosterone[J]. JAMA, 2017, 317(7): 708–716. doi: 10.1001/jama.2016.21043
 Authors and Reviewers
Authors and Reviewers

 DownLoad:
DownLoad:

