The Key Laboratory of Rare Metabolic Disease, Department of Biochemistry and Molecular Biology, the Key Laboratory of Human Functional Genomics of Jiangsu Province, Key Laboratory of Targeted Intervention of Cardiovascular Disease, Collaborative Innovation Center for Cardiovascular Disease Translational Medicine, Nanjing Medical University, Nanjing, Jiangsu 211166, China
2.
Department of General Surgery, the First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu 210029, China
3.
Department of Endocrinology, the First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu 210029, China
Xu Zhang, Department of Biochemistry and Molecular Biology, Nanjing Medical University, Nanjing, Jiangsu 211166, China. Tel: +86-25-86869418, E-mail: zhangxu@njmu.edu.cn
John Zhong Li, Department of Biochemistry and Molecular Biology of Nanjing Medical University, Nanjing, Jiangsu 211166, China. Tel: +86-25-86869427, E-mail: lizhong@njmu.edu.cn
Nonalcoholic fatty liver disease (NAFLD) is considered a major health epidemic with an estimated 32.4% worldwide prevalence. No drugs have yet been approved and therapeutic nodes remain a major unmet need. Long noncoding RNAs are emerging as an important class of novel regulators influencing multiple biological processes and the pathogenesis of NAFLD. Herein, we described a novel long noncoding RNA, lnc_217, which was liver enriched and upregulated in high-fat diet-fed mice, and a genetic animal model of NAFLD. We found that liver specific knockdown of lnc_217 was resistant to high-fat diet-induced hepatic lipid accumulation and decreased serum lipid in mice. Mechanistically, we demonstrated that knockdown of lnc_217 not only decreased de novo lipogenesis by inhibiting sterol regulatory element binding protein-1c cleavage but also increased fatty acid β-oxidation through activation of peroxisome proliferator-activated receptor α and carnitine palmitoyltransferase-1α. Taken together, we conclude that lnc_217 may be a novel regulator of hepatic lipid metabolism and a potential therapeutic target for the treatment of hepatic steatosis and NAFLD-related metabolic disorders.
Nonalcoholic fatty liver disease (NAFLD) has emerged as a significant public health problem, paralleling the dramatic escalation in the global prevalence of obesity. NAFLD affects about 32.4% of people worldwide, with prevalence rates varying from 13% in Africa to 42% in Southeast Asia[1]. By 2030, the number of NAFLD cases in China is predicted to increase to 314.58 million, which is the fastest growth in the prevalence of NAFLD globally[2]. Currently, there are no licensed drug treatments available for NAFLD, although there have been many drugs in the pipeline that are reckoned as good candidates to cure NAFLD/nonalcoholic steatohepatitis[3]. There is an unmet need to use natural products to cure or alleviate NAFLD by physicians[4].
The pathophysiology of NAFLD has not been elucidated. However, it is known to develop when the influx of lipids traveling into the liver (i.e., fatty acid uptake and de novo lipogenesis [DNL]) exceeds hepatic lipid disposal (i.e., mitochondrial fatty acid oxidation and exportation as a component of very low-density lipoprotein [VLDL] particles). Studies reported that hepatic DNL made up about one-third of the total triglyceride content in the liver of NAFLD patients with hyperinsulinemia[5–6]. Sterol regulatory element binding protein-1c (SREBP-1c) is a key lipogenic transcription factor, which directly activates the expression of more than 30 proteins, including fatty acid synthase (FAS) and acetyl-CoA carboxylase (ACC), and is involved in fatty acid and triglyceride synthesis[7]. Several lines of evidence implied that hyperinsulinemia, which predominated in the insulin-resistant state, stimulated lipogenesis by activating SREBP-1c, causing NAFLD in humans and animal models[8–9]. During starvation, the body is powered mainly by adenosine triphosphate through β-oxidation of fatty acids, and this process was found to be modulated by carnitine palmitoyltransferase-1A (CPT-1A), a rate-limiting enzyme playing a crucial role in controlling fatty acyl-coA shuttle from the cytosol into mitochondrial matrix, where fatty acyl-coA underwent β-oxidation to produce energy in the form of adenosine triphosphate[10]. Moreover, transcription factor peroxisome proliferator-activated receptor α (PPARα) was reported to promote the transcription of CPT-1 to activate fatty acid β-oxidation and the generation of ketone bodies[11]. During starvation or fasting, one study showed that the defects in the Pparα gene led to hepatic steatosis and the development of hypoketosis and hypoglycemia in mice[12]. Of note, in previous studies, SREBP-1c expression decreased in PPARα-null mice, compared with wild-type mice[13], and PPARα agonists enhanced the activity of the SREBP-1c promoter through direct binding with its DR1 motif[14]. Others reported that malonyl-CoA produced by ACC inhibited the activity of CPT-1, and thereby decreased the rate of β-oxidation by reducing fatty acid transport to mitochondria[15–16].
Long non-coding RNAs (lncRNAs) are a type of RNA, generally defined as transcripts more than 200 nucleotides that are not translated into protein[17–18]. Similar to protein-coding genes, many lncRNAs are restricted in the tissue distribution[19]. Recent studies have demonstrated that lncRNAs are essential for fatty acid biosynthesis, oxidation, and VLDL secretion in the liver[20–22]. For example, lncHR1 was reported to be involved in the activation of SREBP-1c to regulate hepatic fatty acid synthesis[23]. Up-regulated long non-coding RNA (HULC) was found to target PPARα to regulate lipid deposition in hepatocellular carcinoma[24]. Therefore, to further identify new lncRNAs that regulate hepatic lipid metabolism is of great importance.
To explore new non-coding RNA related to liver lipid metabolism, we analyzed an existing RNA-seq dataset (GSE157482) from the livers of high-fat diet (HFD) and normal diet (ND) mice and identified a novel lipid-induced lncRNA, lnc_217. To investigate its function, we performed gain and loss of function experiments to manipulate lnc_217 expression levels in vivo and in vitro, based on adenovirus transduction and plasmid transfection, to clarify the role of a novel lncRNA in hepatic fatty acid metabolism, and to provide a new potential therapeutic target for the treatment of hepatic steatosis and NAFLD-related metabolic disorders.
Materials and methods
Experimental animals
C57BL/6J mice used in the current study were purchased from the Model Animal Research Center of Nanjing University (MARC, Nanjing, Jiangsu, China). All of the mice were maintained in a 12-h light/dark cycle at 25 ℃ with free access to rodent chow and water in the animal facility (specific-pathogen free) of Nanjing Medical University. All the procedures followed the guidelines for the care and use of animals established by Nanjing Medical University (IACUC-1601211).
Cell culture and transfection
293A and Hepa1-6 cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and maintained in high-glucose DMEM (Life Technologies, Gaithersburg, MD, USA) containing 10% FBS (Gibco, Waltham, MA, USA) and 1% penicillin-streptomycin. The full-length lnc_217 expression vectors with Myc-Tag were amplified by PCR from C57BL/6J mouse liver cDNA. The specific primers were listed as follows: lnc_217-F: 5′-GTGCCAGACTACGCAGGATCCATGCTCATCATCTTGCCTCTGGG-3′; lnc_217-R: 5′-CGCCTCGAGAAGCTTGGATCCGTGGCTTTCCTGTGCACGTGTTG-3′; lnc_217-T-R: 5′-CGCCTCGAGAAGCTTGGATCCTGTGGCTTTCCTGTGCACGTGTTG-3′; lnc_217-TT-R: 5′-CGCCTCGAGAAGCTTGGATCCTTGTGGCTTTCCTGTGCACGTGTTG-3′. Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) was used for transient transfection in 293A and Hepa1-6 cells according to the manufacturer's instructions.
Adenovirus-mediated knockdown of lnc_217 in mice
Adenovirus of Ad-scramble or Ad-shlnc_217 was generated by the AdEasy system (shlnc_217 target sequences: 5′-CTAAGCATGACAAATCACT-3′, 3′- AGTGATTTGTCATGCTTAG-5′) and injected (n = 6/group, 2.5 × 1011 particles/mouse) via tail veins of 8–10-week-old C57BL/6J mice. After injection, mice were given either an ND or an HFD (60% kcal from fat, D12492, Research Diets, New Brunswick, NJ, USA) for seven days. After a 16-h fast, the mice were then euthanatized, and the liver and blood were harvested.
Mouse primary hepatocyte isolation and treatment
Mouse primary hepatocytes were isolated in a previously mentioned manner[21]. Hepatocytes were cultured in a low glucose medium (Gibco) with 10% FBS supplement in 6-well plates at a density of 4 × 105 cells per well. Adenovirus was applied to primary hepatocytes (6 × 109 particles/well) for 6 h. Following a 16-h incubation period with or without 200 μmol/L oleic acid, cells were harvested.
Real-time reverse transcription PCR (RT-qPCR)
The livers were immediately frozen in liquid nitrogen, and RNA was isolated using Trizol (Invitrogen), according to the manufacturer's instructions. The cDNA synthesis and quantitative PCR with the indicated primers (Supplementary Table 1, available online) were performed as described. RNA expression levels were normalized to Actb as the internal control and to the control group using the 2−ΔΔCt method, calculated as arbitrary units, and represented as mean ± standard deviation. The data of the expression levels of lnc_217 in different tissues of C57BL/6J mice were expressed as mean.
Western blotting
Fresh tissues or cells were harvested and lysed in RIPA buffer supplemented with a complete protease inhibitor cocktail (Roche Diagnostics Deutschland GmbH, Mannheim, Germany), and their protein concentration was subsequently determined by the BCA assay. Then, SDS-PAGE and wet transfer were performed. The membranes were blocked with 5% milk and then incubated with the indicated primary antibodies followed by HRP-conjugated secondary antibody. Proteins were visualized using Super-Signal ECL (Biotanon, Shanghai, China). The following antigens were targeted by the use of antibodies: anti-SREBP-1c (1∶1000; Cat. #ab28481, Abcam, Cambridge, UK), anti-PPAR (1∶1000; Cat. #ab24509, Abcam), anti-CPT1α (1∶1000; Cat. #CPT1L12-A, Alpha Diagnostic, San Antonio, TX, USA), anti-UCP2 (1∶1000; Cat. #ab67241, Abcam), and Calnexin (1∶3000; Cat. #ADI-SPA-860, ENZO Life Sciences, Farmingdale, NY, USA). Protein levels were normalized to calnexin as the internal control and to the control group.
Liver and plasma lipids analysis
Liver lipids were extracted from 100–200 mg of frozen liver samples using the Folch and Lees' method. Triglyceride, total cholesterol and free fatty acid were measured using enzymatic kits (Wako, Richmond, VA, USA) and normalized to sample weight. Plasma triglyceride, total cholesterol and free fatty acid were measured using the same enzymatic assays.
Oil red O staining
Liver sections were embedded in liquid nitrogen and transversely sectioned into 10-μm frozen sections for oil red O staining. Stained sections were analyzed using ImageJ software (NIH, http://rsb.info.nih.gov/ij/). We opened the raw image and split it into three color-channels with "Split channels". The blue channel, which highlighted its raw oil-red staining, was chosen to set an appropriate threshold. Threshold values were determined empirically by selecting a setting that gave the most accurate binary image for a subset of randomly selected photomicrographs with varying peptides densities. Then, the resulting image was measured by "Area fraction" measurements. Six slices were analyzed per group.
5′ and 3′ rapid amplification of cDNA ends (RACE)
To identify the full-length sequence of lnc_217, the GeneRacer Kit (Invitrogen) was used to perform 5′ and 3′ cDNA terminal rapid amplification tests according to the manufacturer's protocol. For the lnc_217 RACE assay, sequences in the mouse genome were retrieved from the NCBI database using the Basic Local Alignment Search Tool, and confirmed by RT-PCR using forward (5′-GGTTAGGACCCGTCAG-3′) and reverse (5′-CAGTGAGCGAGTCTATTT-3′) primers and sequencing. From the sequencing results, 5′ and 3′ RACE primers were designed. The sequences of gene-specific primers were 5′-ACTCATCATCTTGCCTCTGGGAATC-3′ (5′ RACE) and 5′-GCCTCACTGTTTTGCCTGGTG-3′ (3′ RACE).
RNA fluorescence in situ hybridization (RNA-FISH)
Lnc_217 was detected by RNA-FISH in mouse hepatocytes. Cell slides fixed with 4% paraformaldehyde were treated with several buffers in the RNA-FISH Kit (Ri-boBio, Guangzhou, China). After denaturation at 37 ℃ for 5 min, the probe mixture was hybridized overnight in cell slides at 37 ℃ in the dark. CY3-labeled Locked Nucleic Acid probes targeted lnc_217. After washing, the signal was observed under a confocal microscope.
Statistical analysis
Statistical analysis was performed by GraphPad Prism 7. Results were presented as the mean and standard deviation of at least three independent experiments as indicated in the figure legends. A two-tailed Student's t-test was used to calculate the statistical significance of different groups. A two-way ANOVA was used for statistical analysis in Supplementary Fig. 2 (available online). Statistical results were considered significant when P < 0.05.
Results
Identification of a novel lncRNA implicated in hepatic lipid metabolism
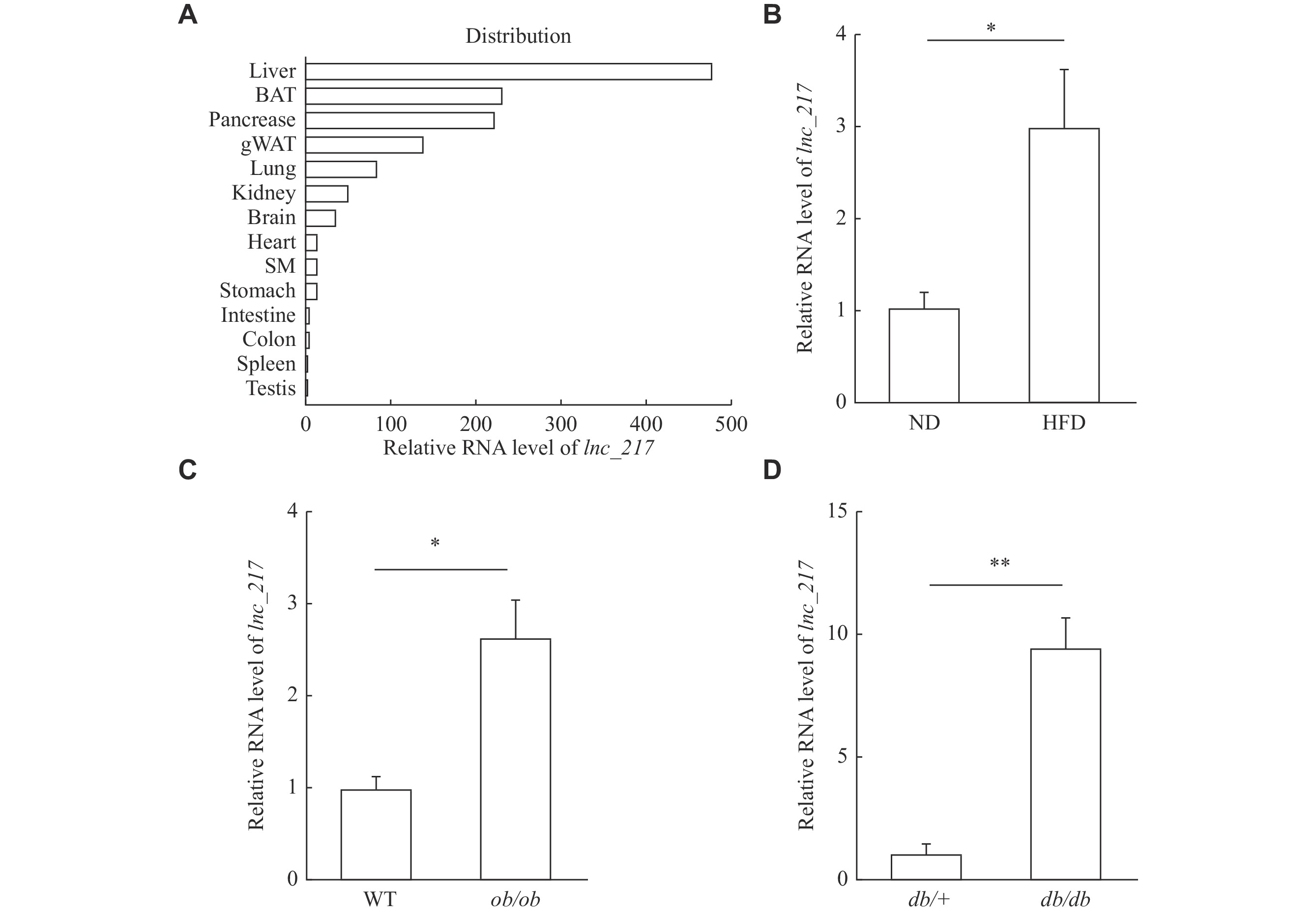
To identify novel lncRNAs potentially involved in hepatic lipid metabolism, we analyzed a previously published RNA-seq dataset (GSE157482) from the livers of HFD and ND mice[21]. Among the altered lncRNAs, we found that the expression of lnc_217 was enriched in the mouse liver (Fig. 1A) and significantly increased in the liver of the HFD mice (Fig. 1B). In addition to the HFD mouse model, ob/ob and db/db mice are important genetic mouse models of NAFLD. The hepatic expression levels of lnc_217 were significantly increased in these two NAFLD mouse models (Fig. 1C–1D). Taken together, lnc_217 is regulated by nutritional status and may be correlated with the pathogenesis of hepatic steatosis.
Figure
1.
Identification of a novel long non-coding RNA (lncRNA) implicated in hepatic lipid metabolism.
A: Relative expression of lnc_217 in mouse tissues (three samples pooled together per tissue). Tissue samples from three C57BL/6J mice were harvested for RNA extraction and pooled for reverse transcription to obtain a set of tissue cDNA. Total RNA was subjected to real-time reverse transcription PCR (RT-qPCR), and the levels of lnc_217 in tissues were expressed relative to the testis. Data are expressed as the mean. B: RT-qPCR analysis of relative lnc_217 expression in the liver of C57BL/6J mice fed with a normal diet (ND) or a high-fat diet (HFD) for four weeks (n = 6/group). C and D: RT-qPCR analysis of relative lnc_217 expression in the livers of ob/ob (C) and db/db (D) mice (n = 6/group). lnc_217 expression levels were normalized to Actb as the internal control and to the control group. Data are expressed as the mean and standard deviation (B–D). Statistical analyses were performed by Student's t-test for two-group comparisons (B–D). *P < 0.05 and **P < 0.01. Abbreviations: BAT, brown adipose tissue; gWAT, white adipose tissue of the groin; SM, skeletal muscle.
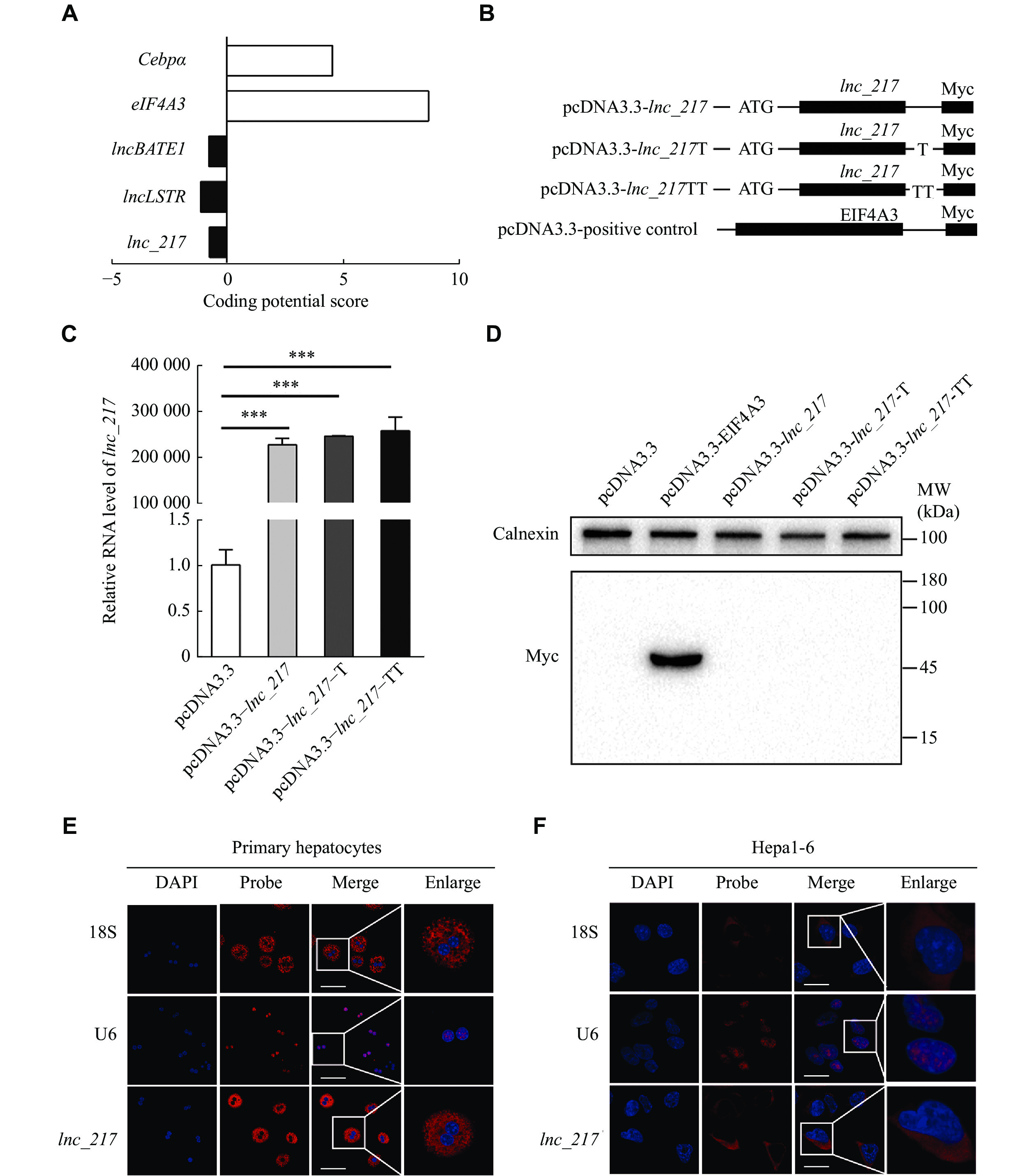
Prediction and verification of the encoding ability of lnc_217
The results of the RACE assay revealed a full-length of 2978 nt for lnc_217 located on chromosome 7 of mice (Supplementary Fig. 1A and 1B, available online), which was highly conserved from mice to humans. According to the prediction of the Coding Potential Calculator online prediction system (http://cpc.gao-lab.org/), lnc_217 lacks coding potential, similar to known lncRNAs, such as lncLSTR and lncBATE1 (Fig. 2A). In addition, the full-length lnc_217 expression vectors with Myc-Tag were constructed and transfected into 293A cells to further elucidate protein coding potential (Fig. 2B). All the vectors were transcribed but failed to produce protein (Fig. 2C and2D). These results confirmed that lnc_217 lacked the ability to encode proteins. RNA-FISH analysis further revealed that lnc_217 was enriched in the cytosol fraction of primary hepatocytes and Hepa1-6 hepatocytes with a similar expression pattern to 18S ribosomal RNA, a positive control marker for the cytosol fraction (Fig. 2E and2F). These results suggest that lnc_217 is a lncRNA localized in the cytosol without the protein coding ability.
Figure
2.
Characterization of the non-coding property and sub-cellular location of lnc_217.
A: Prediction of the coding capacity of lnc_217 using the Coding Potential Calculator. B: The full-length lnc_217 was cloned into the eukaryotic expression vector pcDNA3.3-Myc with an N-terminal codon ATG in the three expression patterns. C: Real-time reverse transcription PCR analysis of the relative expression of Myc-tagged lnc_217. D: Western blotting analysis was performed to detect the expression of Myc-tagged protein, eukaryotic translation initiation factor 4A3 (EIF4A3), with the Myc tag severed as a positive control. E and F: RNA fluorescence in situ hybridization analysis in hepatocytes. Red, lnc_217 or small nuclear (sn) RNA U6 or 18S ribosomal RNA (snRNA U6 and 18S ribosomal RNA were selected as positive controls for the nucleus and cytosol marker, respectively); blue, 4',6-diamidino-2-phenylindole (DAPI) staining. Representative confocal laser microscopy images of primary hepatocytes (E) and Hepa1-6 hepatocytes (F) hybridized with the indicated RNA probes. Scale bar, 60 μm (E) and 20 μm (F). Data are expressed as the mean standard deviation from three independent experiments. Statistical analyses were performed by Student's t-test for two-group comparisons. ***P < 0.001.
Liver-specific knockdown of lnc_217 reduced hepatic lipid accumulation and hyperlipidaemia in HFD-fed mice
To investigate physiological functions of lnc_217 in the hepatic lipid metabolism, we generated liver-specific lnc_217-knockdown mice by tail vein injection with adenoviral-mediated shRNA (Fig. 3A) and then fed the mice an HFD. As shown in Fig. 3B, there was no significant difference in the body weight (Fig. 3B) or the liver index (Fig. 3C) between the lnc_217 knockdown group and the control group. There was also no statistically significant difference in the blood glucose levels between the two groups of mice (Fig. 3D). These findings suggest that liver-specific lnc_217 knockdown may not cause disturbances in the glucose metabolism.
Figure
3.
Knockdown of lnc_217 reduced hepatic lipid accumulation and hyperlidiaemai in high-fat diet fed (HFD) mice.
C57BL/6J male mice (six to eight weeks old) were infected with 2.5 × 1011 recombinant adenovirus particles of Ad-Scramble (Adcon) or Ad-shlnc_217via tail vein injection and fed with a normal diet (ND) or HFD for seven days. Mice were sacrificed and the tissues and plasma were collected for analysis after 16 h fasting (n = 5/group). A: Real-time reverse transcription PCR analysis of the knockdown efficiency of lnc_217 in the liver. lnc_217 expression levels were normalized to Actb as the internal control and to the control group. B: Body weight analysis of the two groups of mice. C: Analysis of the liver-to-body weight ratio of the two groups of mice. D: Comparison of blood glucose in the two groups of mice. E: Representative images of oil red O staining of liver sections from control and lnc_217 knockdown mice (n = 3/group), scale bar, 50 μm. F–H: Total hepatic triglyceride (TG) (F), total cholesterol (TC) (G), and free fatty acid (FFA) (H) were extracted from the livers of Adcon and lnc_217 knockdown (Ad-shlnc_217) mice and measured using enzymatic kits. I–K: Serum TG (I), FFA (J), and TC (K) levels were measured using enzymatic kits. Data are expressed as the mean and standard deviation from three independent experiments. Statistical analyses were performed by Student's t-test for two-group comparisons. *P < 0.05 and **P < 0.01.
To further evaluate biological functions of lnc_217 in the hepatic lipid metabolism, oil red O staining of mouse liver sections was performed. The results showed that the knockdown of lnc_217 significantly inhibited HFD-induced lipid accumulation in the liver (Fig. 3E). Consistently, the hepatic triglyceride level was significantly reduced in liver-specific lnc_217 knockdown mice, compared with that in the control group with HFD challenge (Fig. 3F). However, we found that the knockdown of lnc_217 did not change hepatic cholesterol as well as free fatty acid levels (Fig. 3G and3H). The results of further biochemical analysis of mouse serum revealed that the knockdown of lnc_217 significantly decreased serum triglyceride and free fatty acid levels after HFD feeding (Fig. 3I and3J), without affecting serum total cholesterol levels (Fig. 3K). Taken together, the knockdown of lnc_217 ameliorates HFD-induced liver lipid accumulation and hyperlipidemia in mice.
Liver-specific knockdown of lnc_217 decreased liver fatty acid synthesis and increased β-oxidation
Hepatic lipid homeostasis is mainly coordinated by DNL, fatty acid β-oxidation, and VLDL secretion. We first performed a VLDL-triglyceride secretion assay by injection of tyloxapol to verify the regulatory role of lnc_217 on hepatic lipid secretion in vivo. As shown in Supplementary Fig. 2A (available online), the knockdown of lnc_217 did not alter VLDL secretion.
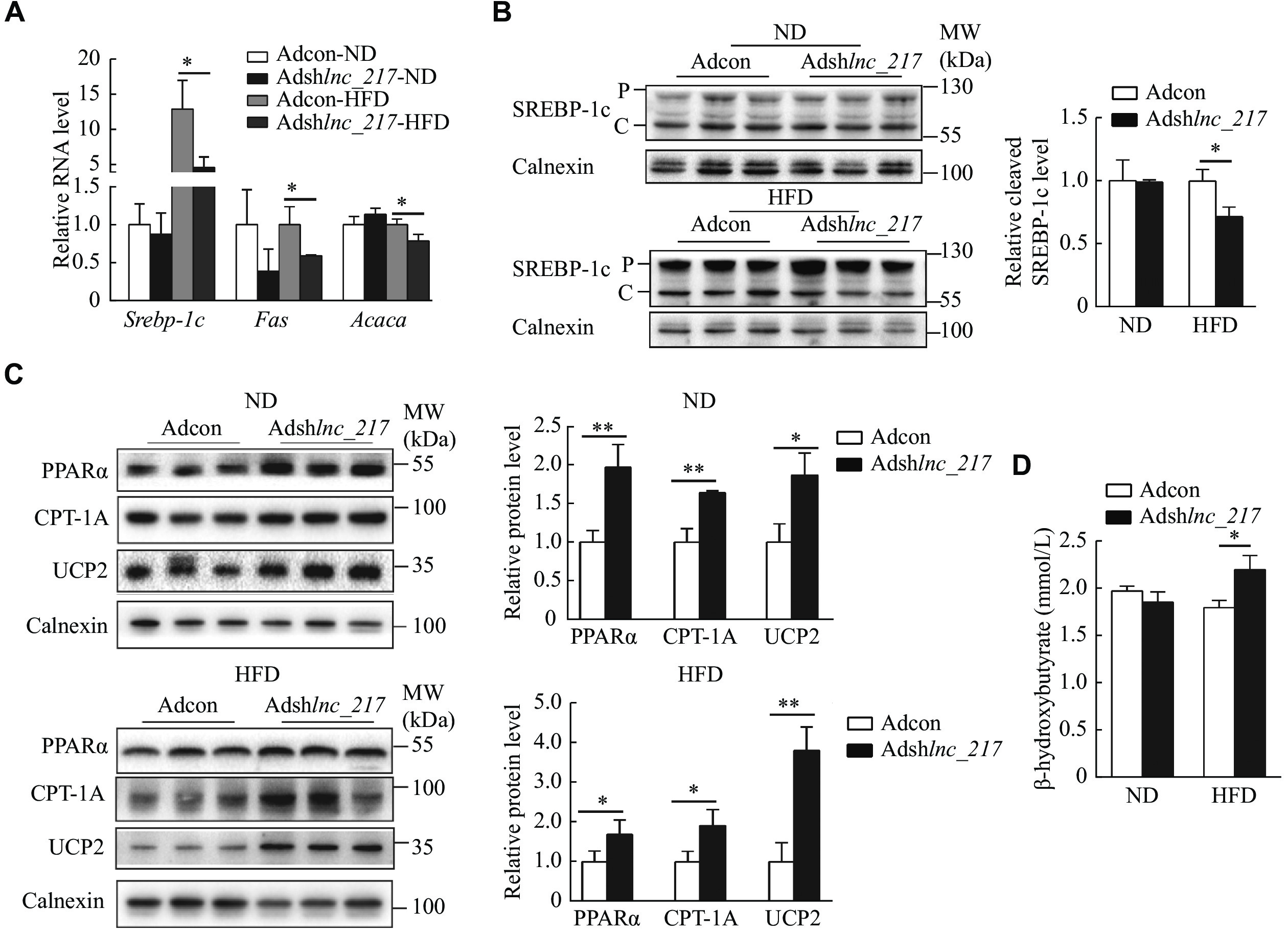
Importantly, the RT-qPCR results showed that liver-specific knockdown of lnc_217 significantly repressed hepatic expression of key genes involved in DNL, such as Srebp-1c, Fas (the gene encoding tumor necrosis factor receptor superfamily member 6), and Acaca (the gene encoding acetyl-CoA carboxylase 1) (Fig. 4A). We further measured the protein level of SREBP-1c in the livers of these mice and found that manipulation of lnc_217 did not change the precursor level of SREBP-1c; however, the cleavage form of SREBP-1c protein level significantly decreased in the liver of lnc_217 knockdown mice with an HFD (Fig. 4B). These results suggest that the knockdown of lnc_217 decreases SREBP-1c expression and cleavage to inhibit hepatic fatty acid synthesis.
Figure
4.
Liver-specific knockdown of lnc_217 decreased hepatic lipogenesis and increased fatty acid β-oxiation in mice.
A: Relative mRNA levels of genes involved in lipogenesis in the livers of Ad-Scramble (Adcon) and lnc_217 knockdown (Ad-shlnc_217) mice under normal diet (ND) or high-fat diet (HFD) conditions (n = 4/group). mRNA levels were normalized to Actb as the internal control and to the control group. B and C: Western blotting analysis of protein levels of SREBP-1c (B) and the of genes involved in β-oxidation (C) from the same livers (n = 3/group). P and C indicate precursor and cleavage forms of SREBP-1c, respectively. Protein levels were normalized to calnexin as the internal control and to the control group. D: Serum ketone body content was measured using enzymatic kits (n = 5/group). Data are expressed as the mean and standard deviation from three independent experiments. Statistical analyses were performed by Student's t-test for two-group comparisons. *P < 0.05 and **P < 0.01. Abbreviations: SREBP-1c, sterol regulatory element binding protein-1c; PPARα, peroxisome proliferator-activated receptor α; CPT-1A, carnitine palmitoyltransferase 1A; UCP2, uncoupling protein 2.
Furthermore, we observed that the protein levels of PPARα, CPT-1A and UCP2 were significantly induced in the liver of mice with the depletion of lnc_217 (Fig. 4C). Consistently, the serum level of β-hydroxybutyrate, a major ketone body generated by fatty acid β-oxidation in the liver, was significantly increased in the lnc_217 knockdown mice after HFD feeding (Fig. 4D). These results suggest that the knockdown of lnc_217 promotes fatty acid β-oxidation in the liver.
Knockdown of lnc_217 decreased fatty acid synthesis and increased β-oxidation in primary hepatocytes
To confirm that the decrease in liver triglyceride in the lnc_217 knockdown mice was cell autonomous, we further investigated biological consequences of the lnc_217 knockdown on the lipid metabolism in primary hepatocytes infected with Ad-shlnc_217 (Fig. 5A). As shown in Fig. 5B, the triglyceride levels were reduced by about 40% in hepatocytes after knockdown of lnc_217 in the presence of oleic acid. Consistent with the observations in mice liver, the mRNA levels of genes involved in lipogenesis, such as Srebp-1c, Fas, Acc1, and Scd1 (the gene encoding acyl-CoA desaturase 1), markedly declined in lnc_217-knockdown hepatocytes with oleic acid treatment (Fig. 5C). The cleavage of SREBP-1c was also reduced (Fig. 5D). In addition, the expressions of PPARα and CPT-1A were significantly upregulated in both mRNA (Fig. 5E) and protein levels (Fig. 5F). These data suggest that the reduced lipid contents in lnc_217 knockdown hepatocytes result directly from a combination effect of the reduced DNL and the increased fatty acid β-oxidation.
Figure
5.
Knockdown of lnc_217 decreased lipogenesis and increased fatty acid β-oxidation in primary hepatocytes.
A: Knockdown efficiency of lnc_217 in Hepa1-6 cells by real-time reverse transcription PCR analysis (n = 3/group). B: Intracellular triglyceride (TG) levels in lnc_217 knockdown primary hepatocytes with or without oleic acid (OA) treatment (n = 3/group). C: Relative mRNA levels of lipogenic genes in lnc_217 knockdown primary hepatocytes with or without OA treatment (n = 3/group). D: Western blotting analysis of protein levels of SREBP-1c in lnc_217 knockdown primary hepatocytes with or without OA treatment (n = 3/group). P and C indicate precursor and cleavage forms of SREBP-1c, respectively. E and F: Relative mRNA (E) and protein (F) levels of genes involved in β-oxidation in lnc_217 knockdown primary hepatocytes with or without OA treatment. mRNA levels were normalized to Actb as the internal control and to the control group. Protein levels were normalized to calnexin as the internal control and to the control group. Data are expressed as the mean and standard deviation from three independent experiments. Statistical analyses were performed by Student's t-test for two-group comparisons. *P < 0.05, **P < 0.01, and ***P < 0.001. Abbreviations: SREBP-1c, sterol regulatory element binding protein-1c; PPARα, peroxisome proliferator-activated receptor α; CPT-1A, carnitine palmitoyltransferase 1A; UCP2, uncoupling protein 2.
Overexpression of lnc_217 increased lipogenesis and decreased fatty acid β-oxidation in Hepa1-6 cells
Apart from the knockdown experiments, we overexpressed lnc_217 in Hepa1-6 cells. In agreement with the knockdown result, the overexpression of lnc_217 resulted in a significant accumulation of triglycerides following oleic acid treatment (Fig. 6A and6B). Furthermore, the mRNA levels of lipogenic genes, including Srebp-1c, Fas, Acc1 and Scd1, markedly increased in the lnc_217-overexpressed cells (Fig. 6C), and the cleavage of SREBP-1c was also elevated (Fig. 6D). In addition, the expression levels of fatty acid oxidation genes (Ppara and Cpt1a) were significantly downregulated (Fig. 6E and6F). These data suggest that the overexpression of lnc_217 may directly induce hepatic steatosis. Based on the results from the knockdown and overexpression experiments, we concluded that lnc_217 regulated the hepatic lipid metabolism by modulating lipogenesis and fatty acid oxidation.
Figure
6.
Overexpression of lnc_217 increased lipogenesis and decreased fatty acid β-oxidation in Hepa1-6 cells.
A: Overexpression efficiency of lnc_217 in Hepa1-6 cells by real-time reverse transcription PCR analysis (n = 3/group). B: Intracellular triglyceride (TG) levels in lnc_217 overexpressed Hepa1-6 cells with or without oleic acid (OA) treatment (n = 6/group). C: Relative mRNA levels of lipogenic genes in lnc_217 overexpressed cells with or without OA treatment (n = 6/group). D: Western blotting analysis of protein levels of SREBP-1c in lnc_217 overexpressed-cells with or without OA treatment (n = 6/group). P and C indicate precursor and cleavage forms of SREBP-1c, respectively. E and F: Relative mRNA (E) and protein (F) levels of genes involved in β-oxidation in lnc_217 overexpressed Hepa1-6 cells with or without OA treatment. mRNA levels were normalized to Actb as the internal control and to the control group. Protein levels were normalized to calnexin as the internal control and to the control group. Data are expressed as the mean and standard deviation from three independent experiments. Statistical analyses were performed by Student's t-test for two-group comparisons. *P < 0.05, **P < 0.01, and***P < 0.001. Abbreviations: SREBP-1c, sterol regulatory element binding protein-1c; PPARα, peroxisome proliferator-activated receptor α; CPT-1A, carnitine palmitoyltransferase 1A; UCP2, uncoupling protein 2.
Given the prevalence of NAFLD, its associated complications, and its lack of effective treatment options, the understanding of hepatic fatty acid metabolism is a vital research priority as we strive to find effective prevention or therapeutic strategies for NAFLD[25]. NAFLD stems from a disequilibrium between hepatic fatty acid influx and disposal. The choice of interventional targets is challenging due to the existence of feedback mechanisms among fatty acid absorption, esterification, oxidation and secretion[26]. LncRNAs are emerging as an important class of novel regulators influencing multiple biological processes and the pathogenesis of metabolic diseases[27–28]. In the current study, we demonstrated that a novel lncRNA, lnc_217, may be a potential therapeutic target for NAFLD, as it effectively balanced the low synthesis and high consumption of fatty acids in the liver.
Various conditions lead to hepatic steatosis in the liver. In the current study, experiments with primary hepatocytes could exclude the uptake of free fatty acids from serum by hepatocytes. A tyloxapol-induced VLDL secretion experiment ruled out triglyceride secretion. It is well known that enhanced activation of SREBP-1c is closely associated with the development of hepatic steatosis and dyslipidemia. The suppression of SREBP-1c cleavage has been proven to be an effective approach to improve fatty liver in mice. The knockdown of lnc_217 reduces cleavage of SREBP-1c, as well as the transcription of its target genes. Thus, the knockdown of lnc_217 decreases hepatic steatosis by inhibiting DNL of fatty acids. On the other hand, lnc_217 knockdown increases the levels of PPARα, CPT-1A and UCP2, which are key regulators of β-oxidation, both in vitro and in vivo. In agreement with this observation, lnc_217 knockdown increases the content of ketone bodies in the blood. It is well known that ACC plays a crucial role in fatty acid oxidation by converting acetyl-CoA into malonyl-CoA, which in turn inhibits the CPT-1 activity and fatty acid transportation into mitochondria, thus reducing oxidation rates. Therefore, the reduced ACC levels may, at least in part, account for the increased fatty acid oxidation and energy expenditure in lnc_217 knockdown mice.
The function of lncRNA is determined by its subcellular localization. In the cytoplasm, lncRNAs regulate the stability and translation of target mRNAs either directly or indirectly by utilizing microRNAs[29]. In gastro-carcinoma cells, MSC-induced lncRNAs HCP5 and MACC1-AS exerted anti-tumour effects by inhibiting the ability of microRNA to activate CPT-1 expression and β-oxidation[22,30]. Within the nucleus, lncRNAs play a role in transcriptional regulation in some ways. Human-specific lncHR1 is located in the nucleus and is involved in the transcriptional inhibition of SREBP-1c[23]. Several nuclear-expressed lncRNAs, such as MALAT1, do not affect SREBP-1c transcription but alter protein shearing[31]. Similar to gene expression, subcellular localization of lncRNAs is a dynamic process[32]. In the current study, we found that lnc_217 was distributed mainly in the cytosol. The knockdown of lnc_217 reduced SREBP-1c transcription and cleavage. In addition, genes involved in β-oxidation were significantly elevated at the transcriptional level. This finding implies that lnc_217 may be involved in the stability and translation of target mRNAs or that the subcellular localization of lnc_217 may be altered. However, the specific mechanism remains unclear. In future studies, we will search for direct target genes of lnc_217 through RNA pull-down and RNA-binding protein immunoprecipitation experiments to better understand the molecular mechanism of lnc_217 in regulating hepatic lipid metabolism and the development of NAFLD.
Taken together, the current study provides the first evidence that lnc_217 is closely involved in the regulation of hepatic lipogenesis and β-oxidation. The expression of lnc_217 is increased in a variety of animal models of fatty liver, suggesting that lnc_217 is likely involved in the progression of NAFLD. The knockdown of lnc_217 ameliorated HFD-induced hepatic steatosis and hyperlipidemia. The dual effects of lnc_217 on fatty acid synthesis and β-oxidation make lnc_217 an appropriate therapeutic target for NAFLD. Therefore, we have revealed a new molecular mechanism of lncRNA regulation of the hepatic fatty acid metabolism, which may pave the way for clinical treatments for NAFLD and dyslipidemia.
Acknowledgments
None.
Fundings
This work was supported by grants from the National Natural Science Foundation of China (Grant Nos. 32130050, 32201064, and 82170838), and the Natural Science Research Project of Universities in Jiangsu Province (Grant No. 21KJB180003).
Riazi K, Azhari H, Charette JH, et al. The prevalence and incidence of NAFLD worldwide: a systematic review and meta-analysis[J]. Lancet Gastroenterol Hepatol, 2022, 7(9): 851–861. doi: 10.1016/S2468-1253(22)00165-0
[2]
Zhou J, Zhou F, Wang W, et al. Epidemiological features of NAFLD from 1999 to 2018 in China[J]. Hepatology, 2020, 71(5): 1851–1864. doi: 10.1002/hep.31150
[3]
Negi CK, Babica P, Bajard L, et al. Insights into the molecular targets and emerging pharmacotherapeutic interventions for nonalcoholic fatty liver disease[J]. Metabolism, 2022, 126: 154925. doi: 10.1016/j.metabol.2021.154925
[4]
Tarantino G, Balsano C, Santini SJ, et al. It is high time physicians thought of natural products for alleviating NAFLD. Is there sufficient evidence to use them?[J]. Int J Mol Sci, 2021, 22(24): 13424. doi: 10.3390/ijms222413424
[5]
Diraison F, Moulin P, Beylot M. Contribution of hepatic de novo lipogenesis and reesterification of plasma non esterified fatty acids to plasma triglyceride synthesis during non-alcoholic fatty liver disease[J]. Diabetes Metab, 2003, 29(5): 478–485. doi: 10.1016/S1262-3636(07)70061-7
[6]
Donnelly KL, Smith CI, Schwarzenberg SJ, et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease[J]. J Clin Invest, 2005, 115(5): 1343–1351. doi: 10.1172/JCI23621
[7]
Batchuluun B, Pinkosky SL, Steinberg GR. Lipogenesis inhibitors: therapeutic opportunities and challenges[J]. Nat Rev Drug Discov, 2022, 21(4): 283–305. doi: 10.1038/s41573-021-00367-2
[8]
Um SH, Frigerio F, Watanabe M, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity[J]. Nature, 2004, 431(7005): 200–205. doi: 10.1038/nature02866
[9]
Yan F, Wang Q, Lu M, et al. Thyrotropin increases hepatic triglyceride content through upregulation of SREBP-1c activity[J]. J Hepatol, 2014, 61(6): 1358–1364. doi: 10.1016/j.jhep.2014.06.037
[10]
Lee K, Kerner J, Hoppel CL. Mitochondrial carnitine palmitoyltransferase 1a (CPT1a) is part of an outer membrane fatty acid transfer complex[J]. J Biol Chem, 2011, 286(29): 25655–25662. doi: 10.1074/jbc.M111.228692
[11]
Gross B, Pawlak M, Lefebvre P, et al. PPARs in obesity-induced T2DM, dyslipidaemia and NAFLD[J]. Nat Rev Endocrinol, 2017, 13(1): 36–49. doi: 10.1038/nrendo.2016.135
[12]
Hu X, Tanaka N, Guo R, et al. PPARα protects against trans-fatty-acid-containing diet-induced steatohepatitis[J]. J Nutr Biochem, 2017, 39: 77–85. doi: 10.1016/j.jnutbio.2016.09.015
[13]
Patel DD, Knight BL, Wiggins D, et al. Disturbances in the normal regulation of SREBP-sensitive genes in PPARα-deficient mice[J]. J Lipid Res, 2001, 42(3): 328–337. doi: 10.1016/S0022-2275(20)31655-2
[14]
Fernandez-Alvarez A, Alvarez MS, Gonzalez R, et al. Human SREBP1c expression in liver is directly regulated by peroxisome proliferator-activated receptor α (PPARα)[J]. J Biol Chem, 2011, 286(24): 21466–21477. doi: 10.1074/jbc.M110.209973
[15]
Mao J, DeMayo FJ, Li H, et al. Liver-specific deletion of acetyl-CoA carboxylase 1 reduces hepatic triglyceride accumulation without affecting glucose homeostasis[J]. Proc Natl Acad Sci U S A, 2006, 103(22): 8552–8557. doi: 10.1073/pnas.0603115103
[16]
Savage DB, Choi CS, Samuel VT, et al. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2[J]. J Clin Invest, 2006, 116(3): 817–824. doi: 10.1172/JCI27300
[17]
Guttman M, Amit I, Garber M, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals[J]. Nature, 2009, 458(7235): 223–227. doi: 10.1038/nature07672
[18]
Kapranov P, Cheng J, Dike S, et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription[J]. Science, 2007, 316(5830): 1484–1488. doi: 10.1126/science.1138341
[19]
Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease[J]. Cell, 2013, 152(6): 1298–1307. doi: 10.1016/j.cell.2013.02.012
[20]
Li P, Ruan X, Yang L, et al. A liver-enriched long non-coding RNA, lncLSTR, regulates systemic lipid metabolism in mice[J]. Cell Metab, 2015, 21(3): 455–467. doi: 10.1016/j.cmet.2015.02.004
[21]
Shen X, Zhang Y, Ji X, et al. Long noncoding RNA lncRHPL regulates hepatic VLDL secretion by modulating hnRNPU/BMAL1/MTTP axis[J]. Diabetes, 2022, 71(9): 1915–1928. doi: 10.2337/db21-1145
[22]
He W, Liang B, Wang C, et al. MSC-regulated lncRNA MACC1-AS1 promotes stemness and chemoresistance through fatty acid oxidation in gastric cancer[J]. Oncogene, 2019, 38(23): 4637–4654. doi: 10.1038/s41388-019-0747-0
[23]
Li D, Cheng M, Niu Y, et al. Identification of a novel human long non-coding RNA that regulates hepatic lipid metabolism by inhibiting SREBP-1c[J]. Int J Biol Sci, 2017, 13(3): 349–357. doi: 10.7150/ijbs.16635
[24]
Cui M, Xiao Z, Wang Y, et al. Long noncoding RNA HULC modulates abnormal lipid metabolism in Hepatoma cells through an miR-9-mediated RXRA signaling pathway[J]. Cancer Res, 2015, 75(5): 846–857. doi: 10.1158/0008-5472.CAN-14-1192
[25]
Mashek DG. Hepatic fatty acid trafficking: multiple forks in the road[J]. Adv Nutr, 2013, 4(6): 697–710. doi: 10.3945/an.113.004648
[26]
Gosis BS, Wada S, Thorsheim C, et al. Inhibition of nonalcoholic fatty liver disease in mice by selective inhibition of mTORC1[J]. Science, 2022, 376(6590): eabf8271. doi: 10.1126/science.abf8271
[27]
van Solingen C, Scacalossi KR, Moore KJ. Long noncoding RNAs in lipid metabolism[J]. Curr Opin Lipidol, 2018, 29(3): 224–232. doi: 10.1097/MOL.0000000000000503
[28]
Duan J, Huang Z, Nice EC, et al. Current advancements and future perspectives of long noncoding RNAs in lipid metabolism and signaling[J]. J Adv Res, 2022, doi: 10.1016/j.jare.2022.08.007.
[29]
Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function[J]. Nat Rev Genet, 2016, 17(1): 47–62. doi: 10.1038/nrg.2015.10
[30]
Chen W, Zhang K, Yang Y, et al. MEF2A-mediated lncRNA HCP5 inhibits gastric cancer progression via MiR-106b-5p/p21 axis[J]. Int J Biol Sci, 2021, 17(2): 623–634. doi: 10.7150/ijbs.55020
[31]
Yan C, Chen J, Chen N. Long noncoding RNA MALAT1 promotes hepatic steatosis and insulin resistance by increasing nuclear SREBP-1c protein stability[J]. Sci Rep, 2016, 6: 22640. doi: 10.1038/srep22640
[32]
Munschauer M, Nguyen CT, Sirokman K, et al. The NORAD lncRNA assembles a topoisomerase complex critical for genome stability[J]. Nature, 2018, 561(7721): 132–136. doi: 10.1038/s41586-018-0453-z
 Authors and Reviewers
Authors and Reviewers

 DownLoad:
DownLoad: