Phosphatidylcholine (PC) and phosphatidylethanolamine (PE), which make up the bulk of mammalian cell membrane phospholipids, are recognized for their importance in metabolic health. Perturbations in the ratio of PC:PE can affect membrane integrity and function, which thus have serious health consequences. Imbalance in the hepatic PC and PE membrane content can be linked to metabolic disturbances such as ER stress, fatty liver and insulin resistance. Given that impaired insulin sensitivity underlies the pathology of many metabolic disorders and skeletal muscle is a significant regulator of energy metabolism, it is likely that aberrant phospholipid metabolism in skeletal muscle affects whole-body insulin sensitivity. Sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) activity and mitochondrial function respond to alterations in PC:PE ratio and are associated with glucose homeostasis. Moreover, PC and PE content within the mitochondrial membrane influence mitochondrial respiration and biogenesis and thus, metabolic function. As skeletal muscle phospholipids respond to stimuli such as diet and exercise, understanding the implications of imbalances in PC:PE ratio is of great importance in the face of the rising epidemic of obesity related diseases. This review will summarize the current state of knowledge signifying the links between skeletal muscle PC:PE ratio and insulin sensitivity with respects to PC and PE metabolism, SERCA activity, mitochondrial function and exercise.
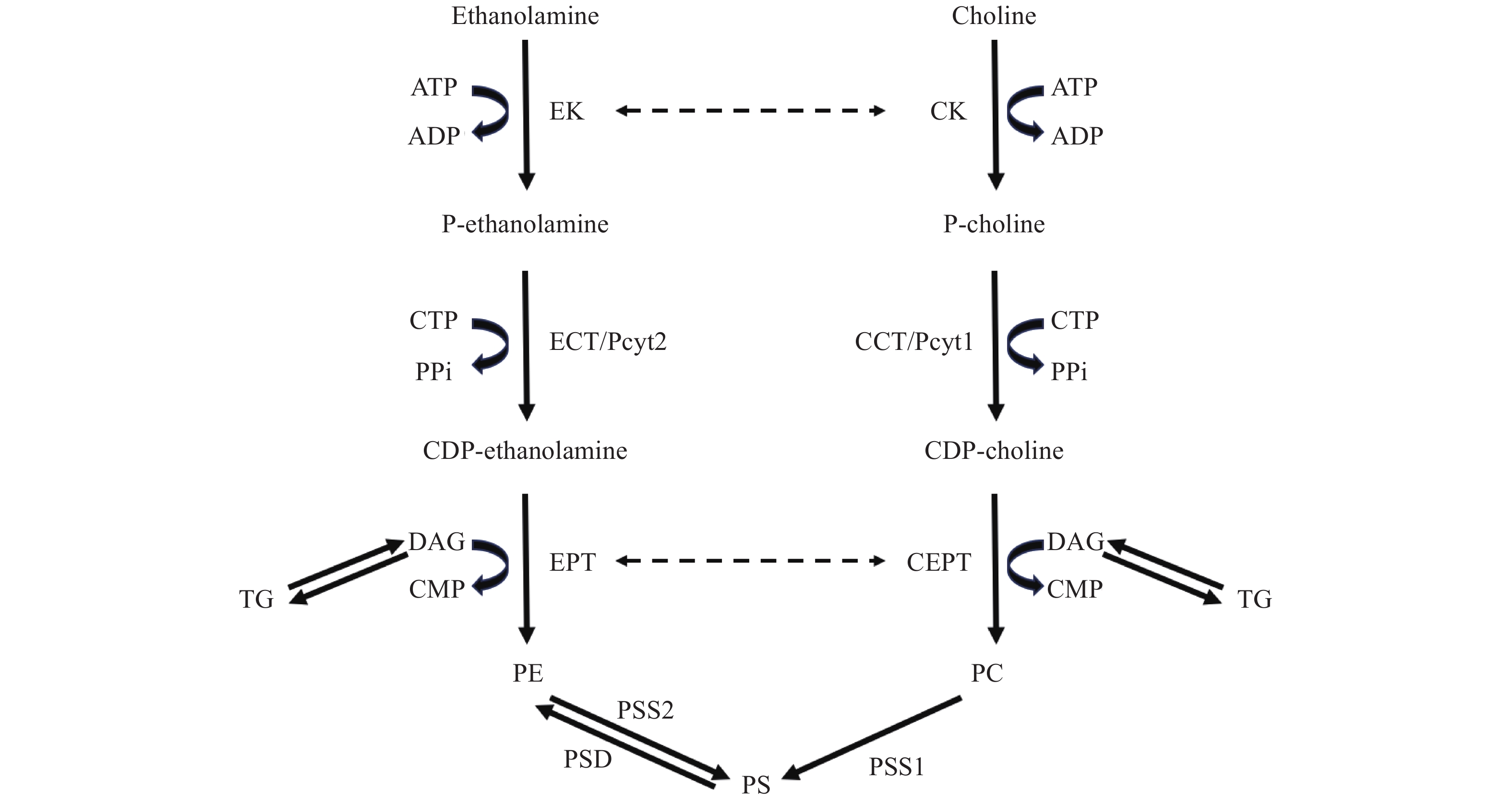
Phosphatidylcholine (PC) and phosphatidylethanolamine (PE) are the most abundant phospholipids in mammalian cell membranes, with PC accounting for 45%–50% and PE for 15%–25% of the total lipid content[1]. PC and PE are synthesized de novo in the analogous cytidine diphosphate (CDP)-choline and CDP-ethanolamine branches of the Kennedy pathway, respectively[2]. In the PC-Kennedy pathway, exogenous choline is taken up by the cell and is initially phosphorylated by choline kinase (CK) to phosphocholine[2]. In the second step, the rate limiting enzyme CTP:phosphocholine cytidylyltransferase (CCT/Pcyt1) converts phosphocholine to CDP-choline using cytidine diphosphate (CTP)[2]. Finally, CDP-choline:1,2-diacylglycerol cholinephosphotransferase (CEPT) catalyzes the condensation of CDP-choline and diacylglycerol (DAG) to produce PC[2]. PE is produced from the analogous PE-Kennedy pathway from ethanolamine and DAG[2]. Alternatively, PE can be produced from the decarboxylation of phosphatidylserine (PS) via phosphatidylserine decarboxylase (PSD) in the mitochondria[3]. PS is produced from preexisting PC and PE through base exchange reactions via phosphatidylserine synthase 1 (PSS1) and phosphatidylserine synthase 2 (PSS2), respectively, in the mitochondrial associated membranes of the ER[3]. Notably, several enzymes of the Kennedy pathway demonstrate overlap in substrate usage[4]. In addition to its putative function in the phosphorylation of choline, multiple mammalian CK isoforms possess the ability to phosphorylate ethanolamine[5]. Likewise, the isoform ethanolamine kinase 2 (EK2) also shows dual kinase activity[6] and several phototransferases are able to use both CDP-choline and CDP-ethanolamine[7]. However, CCT and CTP:phosphoethanolamine cytidylyltransferase (ECT/Pyct2) are highly specific for their substrates (Fig. 1)[3].
Figure
1.
Biosynthetic pathways for PC and PE production.
The major pathway for PC and PE production occurs at the endoplasmic reticulum (ER) viade novo synthesis through the analogous CDP-choline and CDP-ethanolamine branches of the Kennedy pathway, respectively[2]. In the PC-Kennedy pathway, choline is first phosphorylated by CK to P-choline, which is converted to CDP-choline by the rate limiting enzyme CCT/Pcyt1[2]. Lastly, CEPT catalyzes the formation of PC from CDP-choline and DAG. The CDP-ethanolamine branch of the Kennedy pathway is using a similar set of reactions except for the involvement of ethanolamine instead of choline to form PE[2]. PE can also be produced from PS via PSD in the mitochondria[3]. PS is synthesized from PE by PSS2 or from PC by PSS1 in mitochondria-associated regions of the ER[3]. Dashed lines indicate dual specificity in enzyme substrate usage[5–7]. PE: phosphatidylethanolamine; PC: phosphatidylcholine; PS: phosphatidylserine; CDP: cytidine diphosphate; CTP: cytidine diphosphate; CK: choline kinase; P-choline: phosphocholine; CCT/Pcyt1: CTP:phosphocholine cytidylyltransferase; CEPT: CDP-choline:1,2-diacylglycerol cholinephosphotransferase; DAG: diacylglycerol; TG: triglyceride; EK: ethanolamine kinase; P-ethanolamine: phosphoethanolamine; ECT/Pcyt2: CTP:phosphoethanolamine cytidylyltransferase; EPT: CDP-ethanolamine:1,2-diacylglycerol ethanolaminephosphotransferase; PSD: phosphatidylserine decarboxylase; PSS1: phosphatidylserine synthase 1; PSS2: phosphatidylserine synthase 2.
PC and PE are distributed asymmetrically in the outer and inner leaflets of the plasma membrane: the majority of PC is localized to the outer leaflet, whereas PE is enriched in the inner leaflet[8]. Maintenance of the ratio of PC to PE within cellular membranes is critical for membrane function[9]. Perturbations in this ratio which expose altered levels of PC/PE content to either leaflet of the membrane affect membrane integrity through the alteration of membrane potential and permeability to proteins and cytokines[9–10]. Moreover, these changes in membrane dynamics can affect fluidity of the membrane as well as lipid rafts which in turn can alter insulin receptor kinetics and glucose uptake, indicating the potential ramifications of an altered PC:PE ratio on insulin sensitivity[11–15].
Mounting evidence indicates that PC and PE are key factors in metabolic health[7,16– 17]. Disruptions in hepatic PC:PE ratio due to obesity and its associated oversupply of fatty acids in humans or in gene deletion mouse models being linked to impairments in liver regeneration and the development of varying severities of liver disease[10,18– 21]. When phosphatidylethanolamine N-methyltransferase (PEMT) knockout mice were fed a choline-deficient diet, they showed reductions in the PC:PE ratio with concomitant development of steatosis, steatohepatitis and death from liver failure[10,20]. On the other hand, obese mice showed an increased hepatic PC:PE ratio compared to lean mice, which associated with endoplasmic reticulum (ER) stress and steatosis[19]. Correction of this ratio reduced ER stress and improved glucose homeostasis[19]. Thus, it appears that imbalances in the PC:PE ratio in either direction can be detrimental.
While evidence of the effects of altered PC:PE ratio is apparent in hepatic tissues, less appreciated is phospholipid metabolism in skeletal muscle. Skeletal muscle is intrinsically linked to whole body energy metabolism via the major role it plays in lipid and glucose oxidation[22]. Importantly, skeletal muscle significantly regulates whole-body glucose homeostasis through its large contribution to insulin-stimulated glucose disposal[23]. In obesity, aberrant intramuscular lipid metabolism adversely impacts insulin signaling and consequentially, skeletal muscle insulin resistance developments[24– 25]. The associations between altered hepatic PC:PE ratio and obese phenotype raise the possibility that similar alterations in PC and PE metabolism in skeletal muscle may be occurring given its intimate link with insulin sensitivity. The connection between skeletal muscle insulin resistance and metabolic disorders[24] together with the findings that correction of hepatic PC:PE ratio improves glucose homeostasis, which indicates the likelihood that skeletal muscle PC:PE ratio plays a role in insulin sensitivity[19].
Phosphatidylcholine and phosphatidylethanolamine effects on metabolic dysfunction and insulin sensitivity
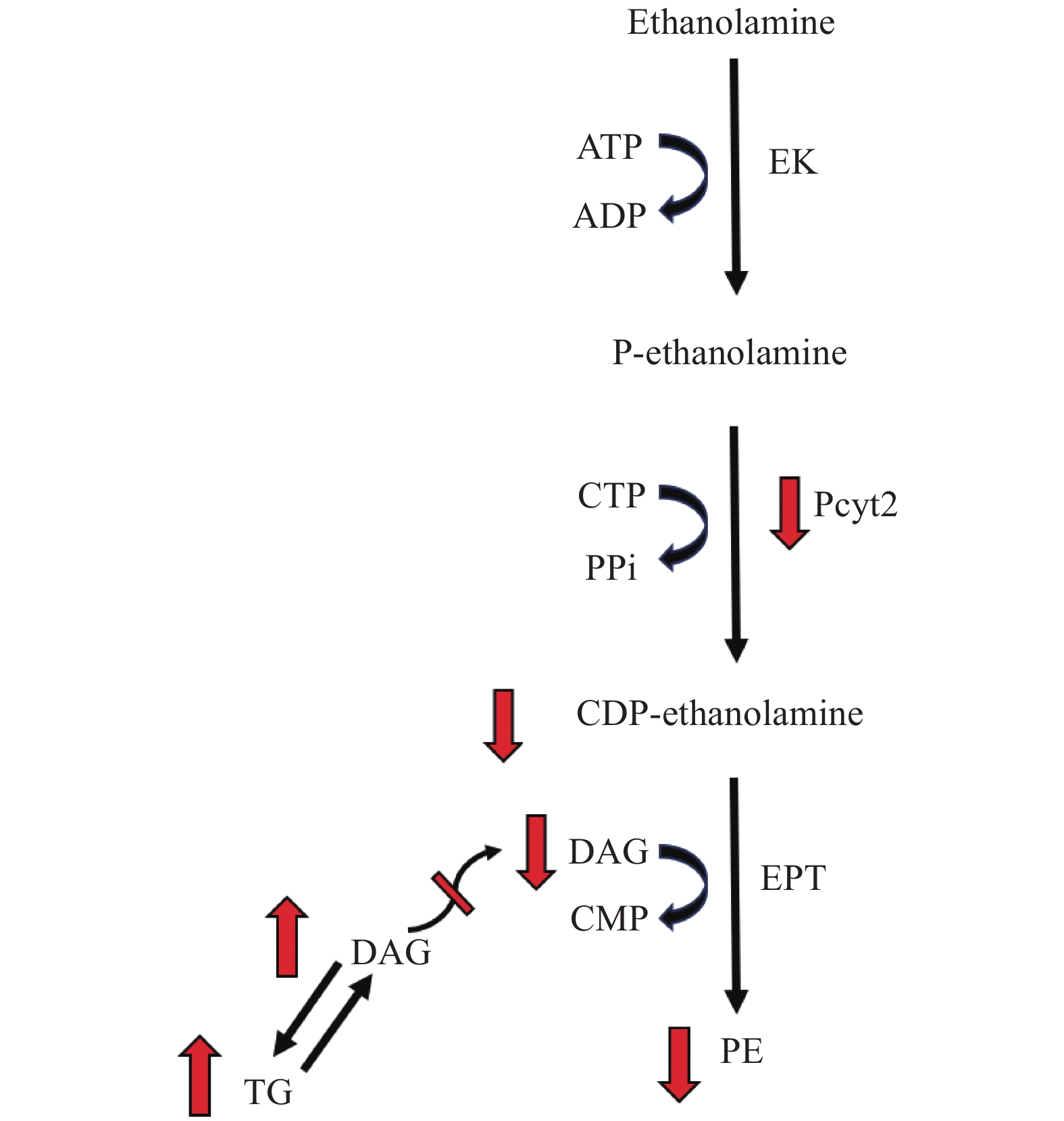
The fundamental importance of phospholipid metabolism in growth, development and metabolic health is demonstrated in the serious health consequences associated with deficiencies in pathways for PC and PE synthesis[7,16– 17]. In muscle cells, choline deficiency alters PC homeostasis through a decrease in PC production via downregulation of Pcyt1, the rate limiting enzyme responsible for de novo PC production in the CDP-choline branch of the Kennedy pathway[26]. These cells exhibited fat accumulation in the form of an increase in total triglyceride (TG) content and lipid droplet formation[26]. While deletion of Pcyt2, the rate limiting enzyme in the CDP-ethanolamine pathway, is embryonically lethal[16], Pcyt2 heterozygous mice survive, however, showed reduced flux through the CDP-ethanolamine pathway which limits the rate of PE synthesis[7]. As a substrate, DAG availability has the capacity to regulate flux through the CDP-ethanolamine pathway[27–29]. Reciprocally, flux through the pathway influences DAG accumulation, and thus, TG metabolism as excess DAG is redirected from PE to TG synthesis (Fig. 2)[7,30– 31]. In accordance with evidence from Pcyt1 inhibition in macrophages[30] and hepatocyte Pcyt2 knockdown[32] and knockout[31], skeletal muscle from global heterozygous Pcyt2 (Pcyt2+/−) mice showed elevated DAG and TG content[7]. This occurs as a consequence of decreased DAG utilization in the CDP-ethanolamine pathway and subsequent shift from PE to TG synthesis and along with enhanced expression of several lipogenic genes[7]. Pcyt2+/− mice exhibit a decrease in markers of mitochondrial biogenesis, a downregulation of mitochondrial fatty acid (FA) β-oxidation genes accompanied by a decreased ability to oxidize FA, and develop insulin resistance[7]. These shifts in metabolism lead to hypertriglyceridemia, liver steatosis and adult-onset obesity[7].
Figure
2.
Pcyt2 knockdown redirects DAG from PE to TG synthesis.
The suppression of Pcyt2 activity results in decreased flux through the CDP-ethanolamine pathway and thus, CDP-ethanolamine production is reduced. Inadequate CDP-ethanolamine pools limit the ability of EPT to use of DAG as a substrate in the final reaction of PE production and causes DAG to accumulate in the cell. Consequently, DAG is shifted from a substrate for PE to TG synthesis, leading to increased production and accumulation of TG. EK: ethanolamine kinase; P-ethanolamine: phosphoethanolamine; ECT/Pcyt2: CTP:phosphoethanolamine cytidylyltransferase; EPT: CDP-ethanolamine:1,2-diacylglycerol ethanolaminephosphotransferase; DAG: diacylglycerol; TG: triglyceride.
Interestingly, in contrast to global Pcyt2+/− mice[7], mice with muscle-specific knockout of Pcyt2 (MPcyt2−/−) showed increased muscle mitochondrial biogenesis, oxidative capacity and exercise performance[15]. MPcyt2−/− mice retained whole body and skeletal muscle insulin sensitivity despite elevations in muscle DAG and TG content[15]. In contradiction with the prevailing hypothesis that suggests the accumulation of DAG causes insulin resistance via activation of PKCθ[33– 34], elevations in muscle DAG did not alter membrane bound PKCθ nor its phosphorylation status; as such, insulin sensitivity was not impacted[15]. The reason for the discrepancy between Pcyt2+/− and MPcyt2−/− is not clear but may be related to altered energy metabolism in other tissues of Pcyt2+/− mice such as the liver[7] or MPcyt2−/− upregulation of FA oxidation[15]. Additionally, skeletal muscle overexpression of Acyl CoA thioesterase 7 (Acot7) in chow fed rats, which, in the tibialis muscle modified PC and PE to levels that resemble those of animals on a high fat diet, induced mixed responses in insulin mediated glucose uptake and β-oxidation across tibialis, red extensor digitorum longus (EDL), white ELD and quadriceps muscles[35]. Thus, distinct characteristics of specific skeletal muscles may also be responsible for the differences in insulin action responses[35]. Moreover, given that Pcyt2 knockout is embryonically lethal[16], a muscle specific knockout model exemplifies an extreme condition and thus, may not accurately represent a likely outcome in a natural model. Nevertheless, together these findings indicate the potential significance of PC and PE homeostasis in metabolic health and importantly, demonstrate their modulatory relationship with insulin sensitivity.
The link between skeletal muscle phospholipid acyl chain composition and insulin sensitivity in humans has been known for over two decades[36– 38]. Briefly, insulin sensitivity has shown to be positively associated with unsaturated fatty acids with the mechanism of action suggested to be due to alterations in insulin receptor concentration and receptor affinity, improved membrane fluidity and membrane-protein dynamics[13,39–40]. However, the relationships between the different classes of phospholipids and insulin sensitivity are less clear. While it is evident that alterations in PC and PE synthetic pathways affect metabolic health, the levels and ratio of PC:PE in skeletal muscle and its association to insulin action is more recently being investigated.
In agreement with findings from human primary myocytes[41], recent evidence from human studies indicates that skeletal muscle PC:PE ratio is inversely associated with insulin sensitivity[42–43]. In endurance trained athletes (ATH), obese sedentary adults (OB) and type 2 diabetics (T2D), basal skeletal muscle PC:PE ratio was negatively correlated with insulin sensitivity while total levels of PE and PC are positively related to insulin sensitivity[42]. Moreover, this association occurred across all participants and in a group specific manner where the ATH group exhibited lower PC:PE ratio and higher insulin sensitivity compared to T2D, with similar trends for OB[42]. Similarly, in both normal and dysglycemic men, basal PC:PE ratio was shown to negatively correlate with insulin sensitivity across all participants[43]. However, these findings are contradicted by animal models that demonstrate no effect on insulin action despite altered PC and PE content. Reduced levels of PC and PE in rat tibialis muscle[35], and PE in mouse skeletal muscle[15] did not influence insulin sensitivity. Moreover, while correction of obesity-induced alterations in hepatic PC:PE ratio improved insulin sensitivity in mice, levels of other lipid species and their fatty acid composition were also modified[19]. Thus, the influence of phospholipid content on insulin action cannot be solely attributed to the alteration of PC:PE ratio and may also or instead involve changes in other major lipid species and their fatty acid tails. Whether skeletal muscle PC:PE ratio plays a direct role in insulin sensitively is unclear; however, these findings suggest possible associations between PC and PE content and insulin resistance that warrant further investigation. There are several links between PC:PE ratio and insulin sensitivity namely, sarco/endoplasmic reticulum Ca2+ ATPase(SERCA) activity and mitochondrial function, which will be explored below.
Sarco/endoplasmic reticulum Ca2+ ATPase
The SERCA is a multi-domain membrane-spanning protein that regulates muscle contractile function by inducing muscle relaxation through the translocation of Ca2+ from the cytosol into the lumen of the sarcoplasmic reticulum (SR)[44]. The ability of lipid bilayer composition to modulate the catalytic activity of SERCA is well established in vitro[44– 46]. In reconstituted dioleoyl-PC:dioleoyl-PE bilayers, elevations in PE content, and thereby decreases in PC:PE ratio of the membrane system, results in an increase in Vmax of SERCA[44– 46] and a reduction in KCa, i.e., increases the Ca2+ binding affinity of SERCA[44]. On the other hand, increased PC content in the membrane has been found to inhibit the calcium transport activity of SERCA[19,47– 48]. Together, these findings reveal the activating role of PE on SERCA function and demonstrate an inverse correlation between PC:PE and SERCA activity[44–45]. Moreover, novel developments in X-ray crystallography provide support for the key role of phospholipids in SERCA function[49].
The relationship between PC:PE and SERCA function is also reflected in in vivo models. Consistent with the finding that overexpression of Pemt, the PE to PC conversion enzyme, significantly inhibits SERCA activity in liver cells[19], elevated hepatic ER PC:PE ratio in obese mice induces SERCA dysfunction and ER stress[19]. Conversely, reduction in ER PC:PE ratio in obese liver as a result of Pemt suppression via shRNA, significantly improves SERCA function[19], confirming the inhibitory effect of elevated PC:PE in the ER. In muscle, the findings are similar. In overloaded mouse plantaris muscles[50] and a mouse model of muscular dystrophy[51], the SR PC:PE ratio negatively correlates with SERCA activity. The connection between SERCA and insulin sensitivity is established in primary myocytes which show insulin sensitivity and SERCA activity are diminished in response to an elevated SR PC:PE ratio[41]. Additionally, in the liver of obese and diabetic mice SERCA protein and consequently, ER Ca2+ is reduced. Furthermore, overexpression of SERCA in these mice restores glucose tolerance with the likely mechanism being related to an increase in ER chaperone protein function given their preference for high ER Ca2+ levels and thus, enhanced ER folding capacity[52]. Collectively, these findings, together with the associations between PC:PE ratio and insulin sensitivity, suggest that SERCA may provide a mechanistic link in the relationship between PC:PE ratio and insulin resistance. Disruptions in PC:PE ratio of the SR likely influence muscle insulin sensitivity through the disruption of calcium homeostasis[53].
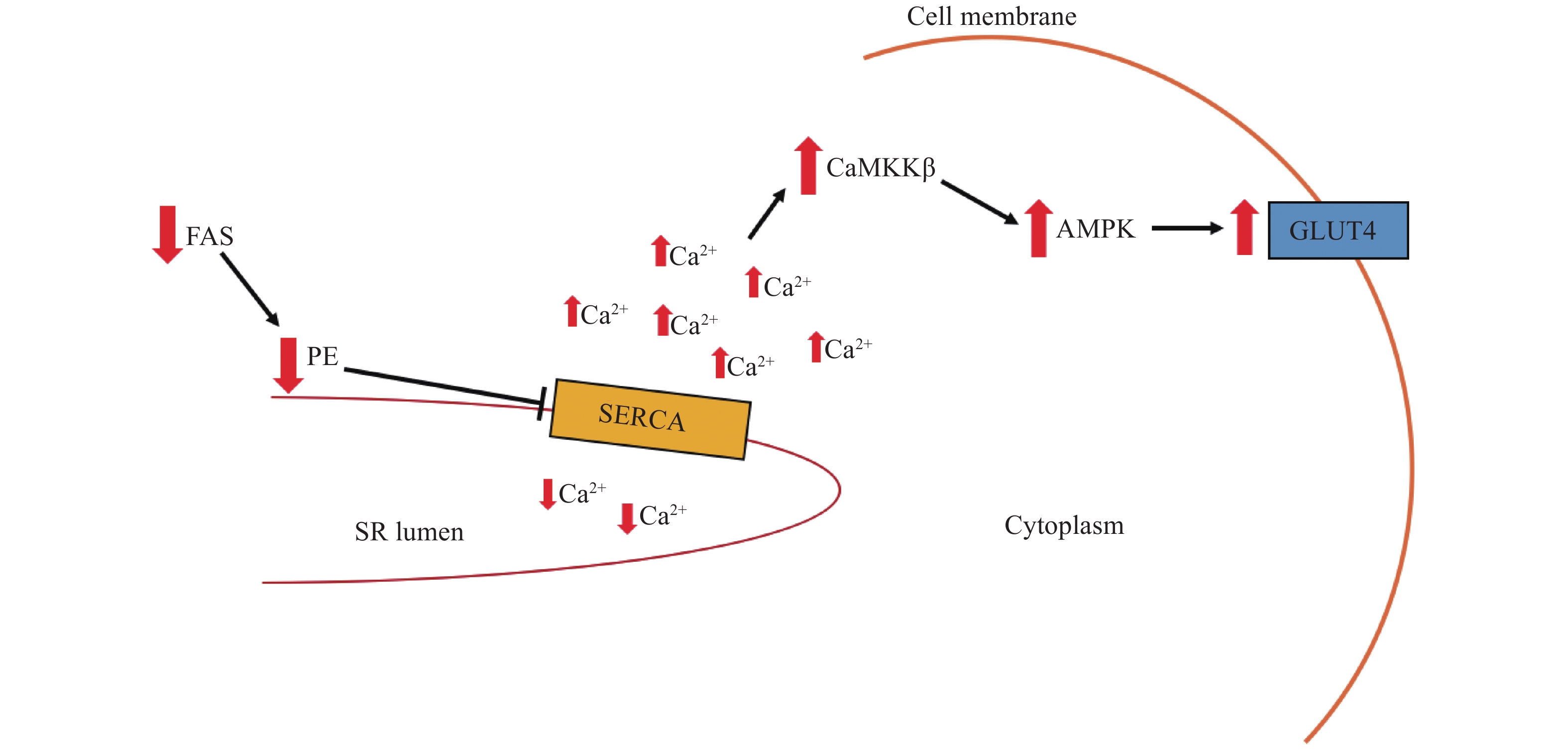
Several recent studies have examined the role of SERCA function in skeletal muscle insulin resistance. In high-fat, high-calorie diets, the enzyme responsible for catalyzing the committed step in de novo lipogenesis, fatty acid synthase (FAS), is suppressed in most tissues[54]. However, the opposite is found to occur in muscle tissues[53]. Elevations in muscle FAS activity in mice with high-fat diet induced obesity is thought to function to increase FAS-facilitated PE synthesis at the SR in order to maintain SERCA activity[53]. This mechanism is supported by the overexpression of FAS in a mouse model of muscular dystrophy, which rescues the impaired SERCA function through an increase in SR PE and subsequent normalization of SR PC:PE ratio[51]. In mice with muscle-specific FAS knockout fed a high-fat diet, PE content is reduced and thus, SERCA activity is inhibited[53]. Unexpectedly, however, this suppression of SERCA lead to improvements in insulin signaling likely due to reduced SERCA mediated Ca2+ uptake and resultant elevations in cytosolic Ca2+ concentration leading to an increase in glucose transport via activation of Ca2+/calmodulin-dependent kinase kinase-β (CaMKKβ) and subsequently, CaMKKβ-dependent 5'-AMP-activated protein kinase (AMPK) phosphorylation and activation[53] (Fig. 3). AMPK stimulates the translocation of GLUT4 to the plasma membrane, enhancing glucose uptake and homeostasis[53]. This finding is corroborated in choline/ethanolamine phosphotransferase 1 (CEPT1) knockdown in C2C12 myoblasts and muscle-specific knockout mice fed a high-fat diet where CEPT1 deficiency increases the SR PC:PE ratio and decreases SERCA activity to preserve insulin sensitivity[14]. However, as proper Ca2+ handling is required for muscle function, both FAS and CEPT1 knockout mice exhibited decreased muscle strength and exercise intolerance, indicating that improved insulin sensitivity due to decreased SERCA activity is likely to come at the cost of appropriate relaxation of muscle fibers and thus, causes muscle weakness[14,53,55]. Lastly, FAS and CEPT knockout mice were protected from diet induced obesity[53], whereas lipid overload in mice fed with a high-fat diet and obese humans is associated with an upregulation of CEPT1 muscle mRNA which is inversely correlated to insulin sensitivity[14]. Thus, it is possible that in vice versa to the finding that CEPT knockdown reduces contractile function at the expense of muscle insulin sensitivity, increased CEPT1 expression induced by high-fat diet feeding and obesity may alter the SR PC:PE to preserve SERCA function and physical performance capacity at the expense of insulin sensitivity[14].
Figure
3.
Mechanism of enhanced insulin sensitivity in FAS deficient mice.
Suppression of FAS, and thus FAS-facilitated PE synthesis, at the SR results in a reduction of SR PE content. Correspondingly, SERCA activity is impaired leading to decreased Ca2+ uptake into the SR lumen and subsequent accumulation of cytosolic Ca2+. CaMKKβ is activated by cytosolic Ca2+ which leads to increased phosphorylation and therefore, activity of AMPK. AMPK activation induces the translocation of GLUT4 to the plasma membrane and thus, enhances insulin-stimulated glucose uptake and improves skeletal muscle glucose homeostasis. FAS: fatty acid synthase; SR: sarcoplasmic reticulum; PE: phosphatidyl ethanolamine; CaMKKβ: Ca2+/calmodulin-dependent kinase kinase-β; AMPK: 5'-AMP-activated protein kinase.
As skeletal muscle mitochondrial respiration significantly contributes to whole body energy metabolism[56], it is hypothesized that deficiencies in skeletal muscle mitochondrial content[57] and function[58] modulate insulin sensitivity. Skeletal muscle mitochondria are dynamic organelles that can readily respond to metabolic stimuli such as exercise[59] or high fat diets[57] through mitochondrial biogenesis and alterations in oxidative capacity. Specifically, mitochondrial mediated alterations in insulin sensitively may be of particular importance in females as the hormone 17β-estradiol (E2) influences mitochondrial membrane fluidity in mouse skeletal muscle[60]. E2 treatment lowered membrane microviscosity and in turn reestablished mitochondrial bioenergetics, respiratory function and insulin sensitivity in E2 deficient female mice, likely as a consequence of the restoration of optimal phospholipid packing and cellular redox homeostasis[60]. These findings indicate the importance of membrane dynamics for mitochondrial function which can be affected by the degree of phospholipid packing and composition[61–62]. Given that mitochondrial phospholipids influence mitochondrial biogenesis[63–64], bilayer-protein interactions[61], the activity of the electron transport system and therefore, mitochondrial function[62,65], skeletal muscle mitochondrial PC:PE ratio may be an important determinant of whole-body insulin sensitivity. Despite this possibility, there is a lack of skeletal muscle mitochondrial phospholipid studies. As such, findings from hepatic studies must be utilized to deduce potential links between alterations PC:PE ratio, mitochondrial activity and insulin sensitivity in skeletal muscle.
The connection between mitochondrial biogenesis, PC and PE, and insulin sensitivity remains largely unclear. The observation that type 2 diabetics and insulin-resistant individuals with impaired glucose tolerance have approximately 30% less skeletal muscle mitochondria than insulin-sensitive individuals[66 – 69], engendered the hypothesis that insulin resistance arises from the accumulation of intramyocellular lipids as a consequence of decreased FA oxidation in mitochondrial deficient skeletal muscle[58,70]. As previously mentioned and in agreement with this hypothesis, MPcyt2−/− mice show increased mitochondrial biogenesis, oxidative capacity, exercise endurance and also demonstrate improved insulin sensitivity[15]. However, this hypothesis is challenged by the findings that in mice fed a high fat diet, insulin resistance still occurs despite an increased mitochondrial content in skeletal muscle[57]. Further, contrary to MPcyt2−/− mice, disruption of the CDP-ethanolamine pathway via muscle specific deletion of CEPT showed no effect on mitochondria, yet, insulin sensitivity improved[14]. The reason for these discrepancies is unclear and highlights the complexity of skeletal muscle mitochondrial PC and PE metabolism.
PE is highly enriched in mitochondrial membranes, containing about 40% of total phospholipids, with the majority of PE being synthesized in situ via mitochondrial phosphatidylserine decarboxylase (PSD)[3,71]. As PE is a nonbilayer-forming phospholipid with a shape that introduces curvature into the mitochondrial membrane, it is specifically enriched in the inner mitochondrial membrane (IMM) to allow for folding of the IMM into crista[72]. The importance of mitochondrial PE production is exemplified in whole body Pisd−/− mice which lacks PSD activity[73]. Despite the continued PE production via CDP-ethanolamine pathway in the ER, PSD knockout is embryonically lethal[73]. The PE deficiency in the mitochondria of these mice causes misshapen, swollen and fragmented mitochondria[73]. Notably, even a moderate <30% mitochondrial PE reduction and corresponding increase in PC:PE ratio in CHO cells induced by Pisd knockdown caused similar aberrant morphology and impaired cell growth[74]. On the other hand, a 33% reduction in PC:PE ratio in hepatic mitochondria of Pemt knockdown mice, due to reduced methylation of PE to PC, results in smaller and more elongated mitochondria[74]. Because oxidative phosphorylation occurs at the inner mitochondrial membrane, proper folding of the membrane is imperative for mitochondrial respiration[72]. In insulin resistance, muscle mitochondria have reported to be smaller and dysfunctional compared to mitochondria from lean volunteers[67], suggesting that PC:PE ratio may influence insulin sensitivity through the regulation of mitochondrial morphology.
PC:PE ratio also appears to be a modulator of mitochondrial oxidative capacity through its influence on mitochondrial respiratory chain (MRC) complexes. In yeast, increased levels of PE have been shown to correlate with biogenesis of MRC components, indicating its role in MRC function[75]. Accordingly, CHO cells with reduced PE content show inhibited activities of electron transport complexes Ⅰ and Ⅳ, resulting in reduced respiration and decreased ATP production[76]. Increased PE content with a corresponding reduction in hepatic PC:PE ratio resulted in an increase in electron transport chain activity, respiration and ATP production with striking correlations between PE and ATP levels[74]. The mechanism underlying these findings remains elusive, however, a specific role for PE in the catalytic activity of MRC complexes is suggested in Arabidopsis thaliana, where depletion of mitochondrial PE decreases complex Ⅳ, and thus respiration, without any effect on individual MRC subunit levels[77]. Moreover, increased mitochondrial PC content in cardiomyocytes reduces electron transport complex Ⅰ, Ⅱ and Ⅳ activity, suggesting an inhibitory effect on enzyme activity[62], possibly through allosteric regulation[61].
PE is further implicated in mitochondrial function as it is a positive regulator[78] and a limiting factor of autophagy[79]. The mitochondrial-selective form of autophagy, mitophagy, is of particular importance in skeletal myocytes given their high metabolic activity which is supported by a large population of mitochondria harbored in intricate networks[80– 81]. As continuous exposure of mitochondria to reactive oxygen species, generated as a by-product of energy production, can cause mitochondrial DNA mutations or perturb protein folding, selective clearance of damaged or dysfunctional mitochondria is imperative to maintain proper function of the mitochondrial network[82]. Additionally, mitophagy is implicated in developmental processes[83– 85] such as the transition from a glycolytic state to one that derives ATP primarily from oxidative phosphorylation in differentiating myoblasts[84– 85]. During this metabolic shift, mitophagy is dramatically upregulated to allow for a dynamic remodeling of the mitochondrial network which involves mitochondrial clearance and subsequent reassembly of functionally different mitochondria capable of robust ATP production[84– 85]. More recently, mitophagy of undamaged mitochondria has been associated with enhanced cellular energy homeostasis through mitochondrial renewal[86–88] which occurs routinely under physiological conditions[86]. In human primary myoblasts[86], and several other cell types[87], mitophagy is dramatically upregulated in response to the stimulation of oxidative phosphorylation[86] and metabolic conditions that favor mitochondrial function and respiration[87] thus indicating mitophagy may routinely enhance energy production through constant regeneration of mitochondria that chronically prevents, rather than to acutely react, to mitochondrial damage. PE is intimately involved in autophagy as it is enriched in the membranes of autophagosomes where it covalently conjugates cytosolic microtubule-associated protein 1 light chain 3 (LC3-Ⅰ) to form the selective autophagosomal protein LC3-Ⅱ[88]. Mitochondrial derived PE, specifically, plays an integral role in the autophagic process. Disruption of mitochondrial/ER connections[89] and direct inhibition of PE formation via PISD knockdown[87] decreases the amplitude of autophagy. Furthermore, the donation of mitochondrial membrane material to autophagosomes formation under basal conditions[90] together with the observation that concurrent elevation of PE levels and autophagy occur in the absence of increased ATP production[87] indicates a role for mitochondrial PE in autophagy for the enhancement of energy production separate from increased MRC activity. Thus, the maintenance of mitochondrial PE levels may influence mitochondrial function and cellular energy homeostasis through its supportive and regulatory role in mitophagy.
Collectively, these findings indicate that the balance of the PC:PE ratio is important for cristae development, mitochondrial shape and growth, can influence mitochondrial respiration and optimize mitochondrial function via mitophagy. Given that reduced muscle mitochondrial function is associated with insulin resistance in skeletal muscle[64–65,91], it is likely that imbalances in the mitochondrial PC:PE ratio can play a modulatory role in muscular insulin sensitivity. Moreover, differences in these studies highlight the complexity of mitochondrial phospholipid metabolism and demand further research as many mechanisms underlying these findings remain unclear.
Response to exercise
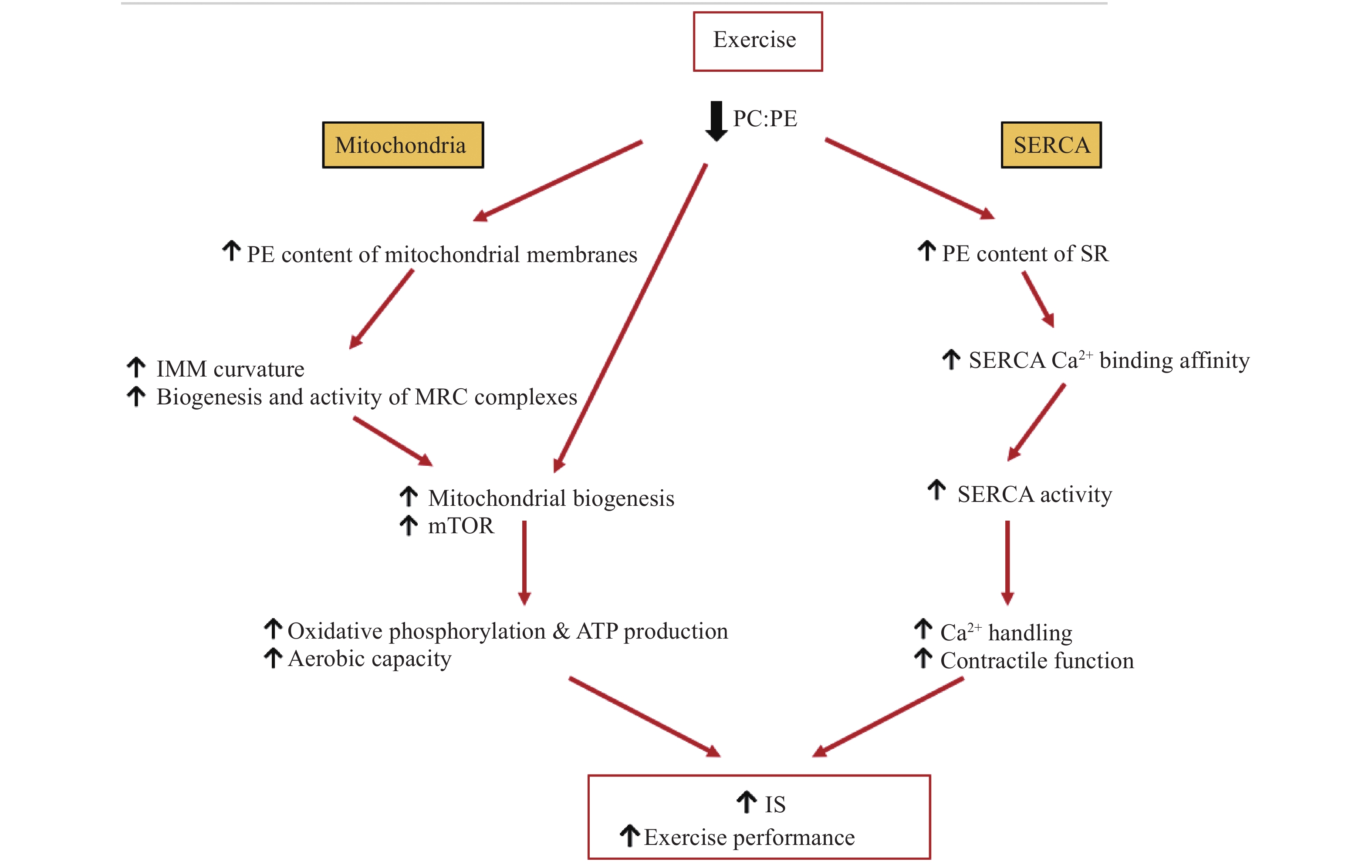
Exercise is known to improve skeletal muscle and whole-body insulin sensitivity and glucose tolerance[92–93] both acutely[94] and chronically[95]. The effect of exercise training on skeletal phospholipid acyl chain composition has been addressed in many studies[96–99], however, less attention has been given to alterations in the skeletal muscle PC:PE ratio in response to exercise. Given the connections between PC:PE ratio and SERCA activity, mitochondrial activity and insulin sensitivity[14–15,64], it is possible that exercise partly exerts its effects on insulin sensitivity through the alteration of PC:PE ratio.
When endurance trained ATH, T2D and OB, were subjected to a single session of exercise, skeletal muscle PC and PE content were markedly altered in a group-specific manner for ATH and T2D; however, PC:PE ratio remained unchanged in all groups[42]. In contrast, a study that measured skeletal muscle PC and PE in normal and dysglycemic men after an acute exercise session (untrained state) and after 12 weeks of exercise intervention (trained state), showed a reduction in the PC:PE ratio after a single exercise session in an untrained state[43]. In these men, 12 weeks of exercise training also resulted in a decreased PC:PE ratio along with an increased total PC and PE levels and improved insulin sensitivity compared to baseline[43]. Interestingly, however, after participants underwent 12 weeks of exercise intervention, an acute exercise session did not alter PC:PE ratio in either group[43]. Moreover, the exercise trained groups from both studies, ATH[39] and normal/dysglycemic men after 12-week training[43], showed a lower PC:PE ratio and greater insulin sensitivity compared to T2D/OB and baseline, respectively[42– 43]. These findings may suggest adaptations to chronic exercise training that favor increased PE and lower skeletal muscle PC:PE ratio to support exercise performance[42–43]. Given that trained men maintain lower PC:PE ratios and improved insulin sensitivity, is possible that in these men a single exercise session does not stimulate the remodeling of membrane PC:PE ratio to the same degree as it does in an untrained state as trained men likely exhibit closer to an optimal PC:PE ratio for exercise performance, and thus, require less alterations to adapt to exercise as untrained men. Of note, even in the absence of exercise training, muscle PC:PE ratio was lower in OB compared to T2D and tended to inversely correlate with insulin sensitivity[43]. This provides evidence that in addition to chronic exercise adaptation, a lowered PC:PE ratio may also support insulin sensitivity independently of exercise. The molecular adaptations to exercise training that link PC:PE ratio and insulin sensitivity may, at least in part, be regulated by SERCA activity and mitochondrial function (Fig. 4). As previously mentioned, reduced PC:PE ratio has an activating effect on SERCA[43], and is associated with insulin sensitive compared to insulin resistant primary myocytes[41]. Conversely, increased PC:PE ratio, impairs SERCA function, negatively impacts exercise performance in FAS and CEPT1 KO mice[14,53] and decreased PE synthesis reduces skeletal muscle mass[14]. As such, it is possible that chronic exercise training may facilitate a decreased PC:PE ratio to support SERCA activity and an increase in insulin sensitivity, skeletal muscle mass, contractile function and exercise capacity. Another possible link between the concomitant exercise-induced alterations in PC:PE ratio and insulin sensitivity relates to mitochondrial activity. In skeletal muscle, insulin resistance is associated with mitochondrial dysfunction[64– 65,91], whereas exercise training increases skeletal muscle mitochondrial density and aerobic capacity[59,100] and alters total skeletal muscle PC/PE content[42– 43]. In normal and dysglycemic men, a decrease in skeletal muscle PC:PE ratio in response to exercise correlated with percent area of mitochondria in muscle cells, skeletal muscle oxidative phosphorylation and mTOR signaling[43], indicating trained groups exhibit increased mitochondrial biogenesis and function. Because mitochondrial membranes are enriched in PE[3], increased mitochondrial biogenesis may help to explain the reduction in overall PC:PE ratio in trained groups. Furthermore, in addition to increased abundance of mitochondria, it is possible that exercise causes a decrease in PC:PE ratio in mitochondrial membranes themselves to support oxidative phosphorylation. Increases in PE in the inner mitochondrial membrane may enhance its folding and therefore facilitate an increase in oxidative capacity[101]. Taken together, these findings may suggest that a lower skeletal muscle PC:PE ratio reflects increases in mitochondrial biogenesis and function as an adaptation to long term exercise. Transcriptomic analysis helps to shed light on the potential mechanisms underlying the alterations in synthesis and degradation pathways for phospholipids that are responsible for modified membrane PC and PE content in response to exercise. After 12 weeks of exercise intervention, mRNA transcripts of CHPT1 and PCYT2 were increased, mirroring the increased PC and PE levels, decreased PC:PE ratio and increased insulin sensitivity[43]. Furthermore, CDP-ethanolamine, the product of ECT (Fig. 1), was shown to be increased both before and after exercise in the more insulin sensitive, ATH group compared to insulin insensitive T2D with a similar trend for ATH compared to OB groups[42]. These findings are in line with a recent genome-wide association study that examined 1 012 human skeletal muscle samples for genes in relation to insulin sensitivity. PCYT2 was found to be positively associated with improvements in insulin sensitively across four independent studies[102]. As ECT catalyzes the rate limiting step in the de novo Kennedy pathway of PE synthesis[103], it is likely that this enzyme plays an important role in insulin sensitivity through its ability to modulate PE production and thus, membrane PC:PE ratio. The upregulation of PCYT2 expression[43] and increase in CDP-ethanolamine[42] in trained compared to untrained groups together with its correlation to insulin sensitivity[42–43,102] suggest that an increase in the enzymatic activity of ECT may reflect an adaptation to chronic exercise which functions to increase insulin sensitivity. These findings together with the metabolic defects observed in Pcyt2 knockdown mice[4,16] suggest the candidacy of ECT/Pcyt2 as a molecular target for therapy given that reduced insulin signaling in skeletal muscle is a hallmark in T2D and obesity related pathologies.
Figure
4.
Potential mechanisms through which exercise-induced decrease in skeletal muscle PC:PE ratio improves insulin sensitivity and exercise performance.
The relationship between skeletal muscle PC:PE ratio and metabolism, and insulin sensitivity is complex. Skeletal muscle PC:PE ratio is negatively correlated with insulin sensitivity, however, whether PC:PE ratio has direct implications for insulin action remains unsolved. It is evident that proper PC:PE ratio is imperative for SERCA function and mitochondrial respiration which both likely contribute to whole body insulin sensitivity through their roles in skeletal muscle; albeit, the mechanisms linking these factors require further clarification. Nevertheless, given that skeletal muscle insulin insensitivity is a hallmark of T2D together with the associations between elevated PC:PE ratio and impaired insulin sensitivity, muscle specific targeting of PC and PE metabolism could provide novel therapeutic options for metabolic disorders.
Acknowledgments
This study was supported by the Canadian Institutes of Health Research grant (CIHR-ECD-144626 Ref # 46309).
Vance JE. Phospholipid synthesis and transport in mammalian cells[J]. Traffic, 2015, 16(1): 1–18. doi: 10.1111/tra.12230
[2]
Kennedy EP, Weiss SB. The function of cytidine coenzymes in the biosynthesis of phospholipides[J]. J Biol Chem, 1956, 222(1): 193–214.
[3]
Schenkel LC, Bakovic M. Formation and regulation of mitochondrial membranes[J]. Int J Cell Biol, 2014, 2014: 709828.
[4]
Fullerton MD, Hakimuddin F, Bonen A, et al. The development of a metabolic disease phenotype in CTP: phosphoethanolamine cytidylyltransferase-deficient mice[J]. J Biol Chem, 2009, 284(38): 25704–25713. doi: 10.1074/jbc.M109.023846
[5]
Aoyama C, Liao HA, Ishidate K. Structure and function of choline kinase isoforms in mammalian cells[J]. Prog Lipid Res, 2004, 43(3): 266–281. doi: 10.1016/j.plipres.2003.12.001
[6]
Lykidis A, Wang JA, Karim MA, et al. Overexpression of a mammalian ethanolamine-specific kinase accelerates the CDP-ethanolamine pathway[J]. J Biol Chem, 2001, 276(3): 2174–2179. doi: 10.1074/jbc.M008794200
[7]
Henneberry AL, McMaster CR. Cloning and expression of a human choline/ethanolamine phosphotransferase: synthesis of phosphatidylcholine and phosphatidylethanolamine[J]. Biochem J, 1999, 339(2): 291–298. doi: 10.1042/bj3390291
[8]
Devaux PF. Static and dynamic lipid asymmetry in cell membranes[J]. Biochemistry, 1991, 30(5): 1163–1173. doi: 10.1021/bi00219a001
[9]
Meikle PJ, Summers SA. Sphingolipids and phospholipids in insulin resistance and related metabolic disorders[J]. Nat Rev Endocrinol, 2017, 13(2): 79–91. doi: 10.1038/nrendo.2016.169
[10]
Li ZY, Agellon LB, Allen TM, et al. The ratio of phosphatidylcholine to phosphatidylethanolamine influences membrane integrity and steatohepatitis[J]. Cell Metab, 2006, 3(5): 321–331. doi: 10.1016/j.cmet.2006.03.007
[11]
Saha S, Anilkumar AA, Mayor S. GPI-anchored protein organization and dynamics at the cell surface[J]. J Lipid Res, 2016, 57(2): 159–175. doi: 10.1194/jlr.R062885
[12]
Górski J, Żendzian-Piotrowska M, de Jong YF, et al. Effect of endurance training on the phospholipid content of skeletal muscles in the rat[J]. Eur J Appl Physiol Occup Physiol, 1999, 79(5): 421–425. doi: 10.1007/s004210050532
[13]
Pilch PF, Thompson PA, Czech MP. Coordinate modulation of D-glucose transport activity and bilayer fluidity in plasma membranes derived from control and insulin-treated adipocytes[J]. Proc Natl Acad Sci USA, 1980, 77(2): 915–918. doi: 10.1073/pnas.77.2.915
Selathurai A, Kowalski GM, Burch ML, et al. The CDP-ethanolamine pathway regulates skeletal muscle diacylglycerol content and mitochondrial biogenesis without altering insulin sensitivity[J]. Cell Metab, 2015, 21(5): 718–730. doi: 10.1016/j.cmet.2015.04.001
[16]
Fullerton MD, Hakimuddin F, Bakovic M. Developmental and metabolic effects of disruption of the mouse CTP: phosphoethanolamine cytidylyltransferase gene (Pcyt2)[J]. Mol Cell Biol, 2007, 27(9): 3327–3336. doi: 10.1128/MCB.01527-06
[17]
Singh RK, Fullerton MD, Vine D, et al. Mechanism of hypertriglyceridemia in CTP: phosphoethanolamine cytidylyltransferase-deficient mice[J]. J Lipid Res, 2012, 53(9): 1811–1822. doi: 10.1194/jlr.M021881
[18]
Ling J, Chaba T, Zhu LF, et al. Hepatic ratio of phosphatidylcholine to phosphatidylethanolamine predicts survival after partial hepatectomy in mice[J]. Hepatology, 2012, 55(4): 1094–1102. doi: 10.1002/hep.24782
[19]
Fu SE, Yang L, Li P, et al. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity[J]. Nature, 2011, 473(7348): 528–531. doi: 10.1038/nature09968
[20]
Walkey CJ, Yu LQ, Agellon LB, et al. Biochemical and evolutionary significance of phospholipid methylation[J]. J Biol Chem, 1998, 273(42): 27043–27046. doi: 10.1074/jbc.273.42.27043
[21]
Martínez-Uña M, Varela-Rey M, Mestre D, et al. S-adenosylmethionine increases circulating very-low density lipoprotein clearance in non-alcoholic fatty liver disease[J]. J Hepatol, 2015, 62(3): 673–681. doi: 10.1016/j.jhep.2014.10.019
[22]
O'Neill HM, Holloway GP, Steinberg GR. AMPK regulation of fatty acid metabolism and mitochondrial biogenesis: implications for obesity[J]. Mol Cell Endocrinol, 2013, 366(2): 135–151. doi: 10.1016/j.mce.2012.06.019
[23]
Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism[J]. Nature, 2001, 414(6865): 799–806. doi: 10.1038/414799a
Steinberg GR. Inflammation in obesity is a common link between defects in fatty acid metabolism and insulin resistance[J]. Cell Cycle, 2007, 6(8): 888–894. doi: 10.4161/cc.6.8.4135
[26]
Michel V, Singh RK, Bakovic M. The impact of choline availability on muscle lipid metabolism[J]. Food Funct, 2011, 2(1): 53–62. doi: 10.1039/C0FO00069H
[27]
Schenkel LC, Sivanesan S, Zhang JZ, et al. Choline supplementation restores substrate balance and alleviates complications of Pcyt2 deficiency[J]. J Nutr Biochem, 2015, 26(11): 1221–1234. doi: 10.1016/j.jnutbio.2015.05.014
[28]
Tijburg LBM, Houweling M, Geelen MJH, et al. Inhibition of phosphatidylethanolamine synthesis by glucagon in isolated rat hepatocytes[J]. Biochem J, 1989, 257(3): 645–650. doi: 10.1042/bj2570645
[29]
Jamil H, Utal AK, Vance DE. Evidence that cyclic AMP-induced inhibition of phosphatidylcholine biosynthesis is caused by a decrease in cellular diacylglycerol levels in cultured rat hepatocytes[J]. J Biol Chem, 1992, 267(3): 1752–1760.
[30]
Jackowski S, Wang JA, Baburina I. Activity of the phosphatidylcholine biosynthetic pathway modulates the distribution of fatty acids into glycerolipids in proliferating cells[J]. Biochim Biophys Acta, 2000, 1483(3): 301–315. doi: 10.1016/S1388-1981(99)00203-6
[31]
Leonardi R, Frank MW, Jackson PD, et al. Elimination of the CDP-ethanolamine pathway disrupts hepatic lipid homeostasis[J]. J Biol Chem, 2009, 284(40): 27077–27089. doi: 10.1074/jbc.M109.031336
[32]
Fullerton MD, Bakovic M. Complementation of the metabolic defect in CTP: phosphoethanolamine cytidylyltransferase (Pcyt2)-deficient primary hepatocytes[J]. Metabolism, 2010, 59(12): 1691–1700. doi: 10.1016/j.metabol.2010.03.022
[33]
Griffin ME, Marcucci MJ, Cline GW, et al. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade[J]. Diabetes, 1999, 48(6): 1270–1274. doi: 10.2337/diabetes.48.6.1270
[34]
Itani SI, Ruderman NB, Schmieder F, et al. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IκB-α[J]. Diabetes, 2002, 51(7): 2005–2011. doi: 10.2337/diabetes.51.7.2005
[35]
Bakshi I, Brown SH, Brandon AE, et al. Increasing Acyl CoA thioesterase activity alters phospholipid profile without effect on insulin action in skeletal muscle of rats[J]. Sci Rep, 2018, 8(1): 13967. doi: 10.1038/s41598-018-32354-w
[36]
Borkman M, Storlien LH, Pan DA, et al. The relation between insulin sensitivity and the fatty-acid composition of skeletal-muscle phospholipids[J]. N Engl J Med, 1993, 328(4): 238–244. doi: 10.1056/NEJM199301283280404
[37]
Pan DA, Lillioja S, Milner MR, et al. Skeletal muscle membrane lipid composition is related to adiposity and insulin action[J]. J Clin Invest, 1995, 96(6): 2802–2808. doi: 10.1172/JCI118350
[38]
Vessby B, Tengblad S, Lithell H. Insulin sensitivity is related to the fatty acid composition of serum lipids and skeletal muscle phospholipids in 70-year-old men[J]. Diabetologia, 1994, 37(10): 1044–1050. doi: 10.1007/BF00400468
[39]
Ginsberg BH, Brown TJ, Simon I, et al. Effect of the membrane lipid environment on the properties of insulin receptors[J]. Diabetes, 1981, 30(9): 773–780. doi: 10.2337/diab.30.9.773
[40]
Nadiv O, Shinitzky M, Manu H, et al. Elevated protein tyrosine phosphatase activity and increased membrane viscosity are associated with impaired activation of the insulin receptor kinase in old rats[J]. Biochem J, 1994, 298(2): 443–450. doi: 10.1042/bj2980443
[41]
Paran CW, Verkerke ARP, Heden TD, et al. Reduced efficiency of sarcolipin‐dependent respiration in myocytes from humans with severe obesity[J]. Obesity, 2015, 23(7): 1440–1449. doi: 10.1002/oby.21123
[42]
Newsom SA, Brozinick JT, Kiseljak-Vassiliades K, et al. Skeletal muscle phosphatidylcholine and phosphatidylethanolamine are related to insulin sensitivity and respond to acute exercise in humans[J]. J Appl Physiol, 2016, 120(11): 1355–1363. doi: 10.1152/japplphysiol.00664.2015
[43]
Lee S, Norheim F, Gulseth HL, et al. Skeletal muscle phosphatidylcholine and phosphatidylethanolamine respond to exercise and influence insulin sensitivity in men[J]. Sci Rep, 2018, 8: 6531. doi: 10.1038/s41598-018-24976-x
[44]
Gustavsson M, Traaseth NJ, Veglia G. Activating and deactivating roles of lipid bilayers on the Ca2+-ATPase/phospholamban complex[J]. Biochemistry, 2011, 50(47): 10367–10374. doi: 10.1021/bi200759y
[45]
Hunter GW, Negash S, Squier TC. Phosphatidylethanolamine modulates Ca-ATPase function and dynamics[J]. Biochemistry, 1999, 38(4): 1356–1364. doi: 10.1021/bi9822224
[46]
Starling AP, Dalton KA, East JM, et al. Effects of phosphatidylethanolamines on the activity of the Ca2+-ATPase of sarcoplasmic reticulum[J]. Biochem J, 1996, 320(1): 309–314. doi: 10.1042/bj3200309
[47]
Li YK, Ge MT, Ciani L, et al. Enrichment of endoplasmic reticulum with cholesterol inhibits sarcoplasmic-endoplasmic reticulum calcium atpase-2b activity in parallel with increased order of membrane lipids[J]. J Biol Chem, 2004, 279(35): 37030–37039. doi: 10.1074/jbc.M405195200
[48]
Cheng KH, Lepock JR, Hui SW, et al. The role of cholesterol in the activity of reconstituted Ca-ATPase vesicles containing unsaturated phosphatidylethanolamine[J]. J Biol Chem, 1986, 261(11): 5081–5087.
[49]
Norimatsu Y, Hasegawa K, Shimizu N, et al. Protein-phospholipid interplay revealed with crystals of a calcium pump[J]. Nature, 2017, 545(7653): 193–198. doi: 10.1038/nature22357
[50]
Fajardo VA, Mikhaeil JS, Leveille CF, et al. Elevated whole muscle phosphatidylcholine: phosphatidylethanolamine ratio coincides with reduced SERCA activity in murine overloaded plantaris muscles[J]. Lipids Health Dis, 2018, 17: 47. doi: 10.1186/s12944-018-0687-7
[51]
Paran CW, Zou K, Ferrara PJ, et al. Lipogenesis mitigates dysregulated sarcoplasmic reticulum calcium uptake in muscular dystrophy[J]. Biochim Biophys Acta, 2015, 1851(12): 1530–1538. doi: 10.1016/j.bbalip.2015.09.001
[52]
Park SW, Zhou YJ, Lee J, et al. Sarco (endo) plasmic reticulum Ca2+-ATPase 2b is a major regulator of endoplasmic reticulum stress and glucose homeostasis in obesity[J]. Proc Natl Acad Sci USA, 2010, 107(45): 19320–19325. doi: 10.1073/pnas.1012044107
[53]
Funai K, Song HW, Yin L, et al. Muscle lipogenesis balances insulin sensitivity and strength through calcium signaling[J]. J Clin Invest, 2013, 123(3): 1229–1240. doi: 10.1172/JCI65726
[54]
Kersten S. Mechanisms of nutritional and hormonal regulation of lipogenesis[J]. EMBO Rep, 2001, 2(4): 282–286. doi: 10.1093/embo-reports/kve071
[55]
Goonasekera SA, Lam CK, Millay DP, et al. Mitigation of muscular dystrophy in mice by SERCA overexpression in skeletal muscle[J]. J Clin Invest, 2011, 121(3): 1044–1052. doi: 10.1172/JCI43844
[56]
Zurlo F, Nemeth PM, Choksi RM, et al. Whole-body energy metabolism and skeletal muscle biochemical characteristics[J]. Metabolism, 1994, 43(4): 481–486. doi: 10.1016/0026-0495(94)90081-7
[57]
Hancock CR, Han DH, Chen M, et al. High-fat diets cause insulin resistance despite an increase in muscle mitochondria[J]. Proc Natl Acad Sci USA, 2008, 105(22): 7815–7820. doi: 10.1073/pnas.0802057105
[58]
Morino K, Petersen KF, Shulman GI. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction[J]. Diabetes, 2006, 55(S2): S9–S15.
[59]
Holloszy JO. Biochemical adaptations in muscle effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle[J]. J Biol Chem, 1967, 242(9): 2278–2282.
[60]
Torres MJ, Kew KA, Ryan TE, et al. 17β-estradiol directly lowers mitochondrial membrane microviscosity and improves bioenergetic function in skeletal muscle[J]. Cell Metab, 2018, 27(1): 167–179. doi: 10.1016/j.cmet.2017.10.003
[61]
Wenz T, Hielscher R, Hellwig P, et al. Role of phospholipids in respiratory cytochrome bc1 complex catalysis and supercomplex formation[J]. Biochim Biophys Acta, 2009, 1787(6): 609–616. doi: 10.1016/j.bbabio.2009.02.012
[62]
Shaikh SR, Sullivan EM, Alleman RJ, et al. Increasing mitochondrial membrane phospholipid content lowers the enzymatic activity of electron transport complexes[J]. Biochemistry, 2014, 53(35): 5589–5591. doi: 10.1021/bi500868g
[63]
Becker T, Horvath SE, Böttinger L, et al. Role of phosphatidylethanolamine in the biogenesis of mitochondrial outer membrane proteins[J]. J Biol Chem, 2013, 288(23): 16451–16459. doi: 10.1074/jbc.M112.442392
[64]
Gohil VM, Greenberg ML. Mitochondrial membrane biogenesis: phospholipids and proteins go hand in hand[J]. J Cell Biol, 2009, 184(4): 469–472. doi: 10.1083/jcb.200901127
[65]
Mejia EM, Hatch GM. Mitochondrial phospholipids: role in mitochondrial function[J]. J Bioenerg Biomembr, 2016, 48(2): 99–112. doi: 10.1007/s10863-015-9601-4
[66]
Petersen KF, Dufour S, Befroy D, et al. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes[J]. N Engl J Med, 2004, 350(7): 664–671. doi: 10.1056/NEJMoa031314
[67]
Kelley DE, He J, Menshikova EV, et al. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes[J]. Diabetes, 2002, 51(10): 2944–2950. doi: 10.2337/diabetes.51.10.2944
[68]
Asmann YW, Stump CS, Short KR, et al. Skeletal muscle mitochondrial functions, mitochondrial DNA copy numbers, and gene transcript profiles in type 2 diabetic and nondiabetic subjects at equal levels of low or high insulin and euglycemia[J]. Diabetes, 2006, 55(12): 3309–3319. doi: 10.2337/db05-1230
[69]
Mootha VK, Lindgren CM, Eriksson KF, et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes[J]. Nat Genet, 2003, 34(3): 267–273. doi: 10.1038/ng1180
[70]
Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes[J]. Science, 2005, 307(5708): 384–387. doi: 10.1126/science.1104343
[71]
Shiao YJ, Lupo G, Vance JE. Evidence that phosphatidylserine is imported into mitochondria via a mitochondria-associated membrane and that the majority of mitochondrial phosphatidylethanolamine is derived from decarboxylation of phosphatidylserine[J]. J Biol Chem, 1995, 270(19): 11190–11198. doi: 10.1074/jbc.270.19.11190
[72]
Basu Ball W, Neff JK, Gohil VM. The role of nonbilayer phospholipids in mitochondrial structure and function[J]. FEBS Lett, 2018, 592(8): 1273–1290. doi: 10.1002/1873-3468.12887
[73]
Steenbergen R, Nanowski TS, Beigneux A, et al. Disruption of the phosphatidylserine decarboxylase gene in mice causes embryonic lethality and mitochondrial defects[J]. J Biol Chem, 2005, 280(48): 40032–40040. doi: 10.1074/jbc.M506510200
[74]
van der Veen JN, Lingrell S, da Silva RP, et al. The concentration of phosphatidylethanolamine in mitochondria can modulate ATP production and glucose metabolism in mice[J]. Diabetes, 2014, 63(8): 2620–2630. doi: 10.2337/db13-0993
[75]
Trotter PJ, Pedretti J, Voelker DR. Phosphatidylserine decarboxylase from Saccharomyces cerevisiae. Isolation of mutants, cloning of the gene, and creation of a null allele[J]. J Biol Chem, 1993, 268(28): 21416–21424.
[76]
Tasseva G, Bai HD, Davidescu M, et al. Phosphatidylethanolamine deficiency in Mammalian mitochondria impairs oxidative phosphorylation and alters mitochondrial morphology[J]. J Biol Chem, 2013, 288(6): 4158–4173. doi: 10.1074/jbc.M112.434183
[77]
Otsuru M, Yu YB, Mizoi J, et al. Mitochondrial phosphatidylethanolamine level modulates cyt c oxidase activity to maintain respiration capacity in Arabidopsis thaliana rosette leaves[J]. Plant Cell Physiol, 2013, 54(10): 1612–1619. doi: 10.1093/pcp/pct104
[78]
Rockenfeller P, Koska M, Pietrocola F, et al. Phosphatidylethanolamine positively regulates autophagy and longevity[J]. Cell Death Differ, 2015, 22(3): 499–508. doi: 10.1038/cdd.2014.219
[79]
Wilson-Zbinden C, dos Santos AXDS, Stoffel-Studer I, et al. Autophagy competes for a common phosphatidylethanolamine pool with major cellular PE-consuming pathways in Saccharomyces cerevisiae[J]. Genetics, 2015, 199(2): 475–485. doi: 10.1534/genetics.114.169797
[80]
Wagatsuma A, Sakuma K. Mitochondria as a potential regulator of myogenesis[J]. Sci World J, 2013, 2013: 593267.
[81]
Leary SC, Battersby BJ, Hansford RG, et al. Interactions between bioenergetics and mitochondrial biogenesis[J]. Biochim Biophys Acta, 1998, 1365(3): 522–530. doi: 10.1016/S0005-2728(98)00105-4
[82]
Pickles S, Vigié P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance[J]. Curr Biol, 2018, 28(4): R170–R185. doi: 10.1016/j.cub.2018.01.004
[83]
Sandoval H, Thiagarajan P, Dasgupta SK, et al. Essential role for Nix in autophagic maturation of erythroid cells[J]. Nature, 2008, 454(7201): 232–235. doi: 10.1038/nature07006
[84]
Fortini P, Iorio E, Dogliotti E, et al. Coordinated metabolic changes and modulation of autophagy during myogenesis[J]. Front Physiol, 2016, 7: 237.
[85]
Sin J, Andres AM, Taylor DJR, et al. Mitophagy is required for mitochondrial biogenesis and myogenic differentiation of C2C12 myoblasts[J]. Autophagy, 2016, 12(2): 369–380. doi: 10.1080/15548627.2015.1115172
Thomas HE, Zhang Y, Stefely JA, et al. Mitochondrial complex I activity is required for maximal autophagy[J]. Cell Rep, 2018, 24(9): 2404–2417. doi: 10.1016/j.celrep.2018.07.101
[88]
Pereira L, Girardi JP, Bakovic M. Forms, crosstalks, and the role of phospholipid biosynthesis in autophagy[J]. Int J Cell Biol, 2012, 2012: 931956.
[89]
Hailey DW, Rambold AS, Satpute-Krishnan P, et al. Mitochondria supply membranes for autophagosome biogenesis during starvation[J]. Cell, 2010, 141(4): 656–667. doi: 10.1016/j.cell.2010.04.009
[90]
Cook KL, Soto-Pantoja DR, Abu-Asab M, et al. Mitochondria directly donate their membrane to form autophagosomes during a novel mechanism of parkin-associated mitophagy[J]. Cell Biosci, 2014, 4(1): 16. doi: 10.1186/2045-3701-4-16
[91]
Koves TR, Ussher JR, Noland RC, et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance[J]. Cell Metab, 2008, 7(1): 45–56. doi: 10.1016/j.cmet.2007.10.013
[92]
Helmrich SP, Ragland DR, Leung RW, et al. Physical activity and reduced occurrence of non-insulin-dependent diabetes mellitus[J]. N Engl J Med, 1991, 325(3): 147–152. doi: 10.1056/NEJM199107183250302
[93]
Soman VR, Koivisto VA, Deibert D, et al. Increased insulin sensitivity and insulin binding to monocytes after physical training[J]. N Engl J Med, 1979, 301(22): 1200–1204. doi: 10.1056/NEJM197911293012203
[94]
Borghouts LB, Keizer HA. Exercise and insulin sensitivity: a review[J]. Int J Sports Med, 2000, 21(1): 1–12. doi: 10.1055/s-2000-8847
[95]
Langleite TM, Jensen J, Norheim F, et al. Insulin sensitivity, body composition and adipose depots following 12 w combined endurance and strength training in dysglycemic and normoglycemic sedentary men[J]. Arch Physiol Biochem, 2016, 122(4): 167–179. doi: 10.1080/13813455.2016.1202985
[96]
Goto-Inoue N, Yamada K, Inagaki A, et al. Lipidomics analysis revealed the phospholipid compositional changes in muscle by chronic exercise and high-fat diet[J]. Sci Rep, 2013, 3: 3267. doi: 10.1038/srep03267
[97]
Senoo N, Miyoshi N, Goto-Inoue N, et al. PGC-1α-mediated changes in phospholipid profiles of exercise-trained skeletal muscle[J]. J Lipid Res, 2015, 56(12): 2286–2296. doi: 10.1194/jlr.M060533
[98]
Mitchell TW, Turner N, Hulbert AJ, et al. Exercise alters the profile of phospholipid molecular species in rat skeletal muscle[J]. J Appl Physiol, 2004, 97(5): 1823–1829. doi: 10.1152/japplphysiol.00344.2004
[99]
Liang MTC, Meneses P, Glonek T, et al. Effects of exercise training and anabolic steroids on plantaris and soleus phospholipids: a 31P nuclear magnetic resonance study[J]. Int J Biochem, 1993, 25(3): 337–347. doi: 10.1016/0020-711X(93)90622-L
[100]
Henriksson J, Reitman JS. Time course of changes in human skeletal muscle succinate dehydrogenase and cytochrome oxidase activities and maximal oxygen uptake with physical activity and inactivity[J]. Acta Physiol Scand, 1977, 99(1): 91–97. doi: 10.1111/j.1748-1716.1977.tb10356.x
Timmons JA, Atherton PJ, Larsson O, et al. A coding and non-coding transcriptomic perspective on the genomics of human metabolic disease[J]. Nucleic Acids Res, 2018, 46(15): 7772–7792. doi: 10.1093/nar/gky570
[103]
Pavlovic Z, Bakovic M. Regulation of phosphatidy-lethanolamine homeostasis—the critical role of CTP: phosphoethanolamine cytidylyltransferase (Pcyt2)[J]. Int J Mol Sci, 2013, 14(2): 2529–2550. doi: 10.3390/ijms14022529
Venkatraman K, Lee CT, Budin I. Setting the curve: the biophysical properties of lipids in mitochondrial form and function. J Lipid Res, 2024, 65(10): 100643.
DOI:10.1016/j.jlr.2024.100643
2.
Gyllenhammer LE, Zaegel V, Duensing AM, et al. Lipidomics of infant mesenchymal stem cells associate with the maternal milieu and child adiposity. JCI Insight, 2024, 9(19): e180016.
DOI:10.1172/jci.insight.180016
3.
Duncan RE. Deficiency of phosphatidylethanolamine synthesis: consequences for skeletal muscle. Function (Oxf), 2023, 4(6): zqad044.
DOI:10.1093/function/zqad044
4.
Grapentine S, Singh RK, Bakovic M. Skeletal Muscle Consequences of Phosphatidylethanolamine Synthesis Deficiency. Function (Oxf), 2023, 4(4): zqad020.
DOI:10.1093/function/zqad020
5.
Zhou E, Wang Q, Li X, et al. Effects of Bee Pollen Derived from Acer mono Maxim. or Phellodendron amurense Rupr. on the Lipid Composition of Royal Jelly Secreted by Honeybees. Foods, 2023, 12(3): 625.
DOI:10.3390/foods12030625
 Authors and Reviewers
Authors and Reviewers

 DownLoad:
DownLoad: