Brianna Lide, Shane McGuire, Hong Liu, Cristina Chandler. Mast cell activation syndrome—anesthetic challenges in two different clinical scenarios[J]. The Journal of Biomedical Research, 2022, 36(6): 435-439. DOI: 10.7555/JBR.36.20220071
Citation:
Brianna Lide, Shane McGuire, Hong Liu, Cristina Chandler. Mast cell activation syndrome—anesthetic challenges in two different clinical scenarios[J]. The Journal of Biomedical Research, 2022, 36(6): 435-439. DOI: 10.7555/JBR.36.20220071
Brianna Lide, Shane McGuire, Hong Liu, Cristina Chandler. Mast cell activation syndrome—anesthetic challenges in two different clinical scenarios[J]. The Journal of Biomedical Research, 2022, 36(6): 435-439. DOI: 10.7555/JBR.36.20220071
Citation:
Brianna Lide, Shane McGuire, Hong Liu, Cristina Chandler. Mast cell activation syndrome—anesthetic challenges in two different clinical scenarios[J]. The Journal of Biomedical Research, 2022, 36(6): 435-439. DOI: 10.7555/JBR.36.20220071
Cristina Chandler and Hong Liu, Department of Anesthesiology and Pain Medicine, University of California Davis Health, 4150 V Street, Suite 1200, Sacramento, CA 95817, USA. Tel/Fax: +1-916-734-5031/+1-916-734-7980, E-mails: cmchandler@ucdavis.edu and hualiu@ucdavis.edu
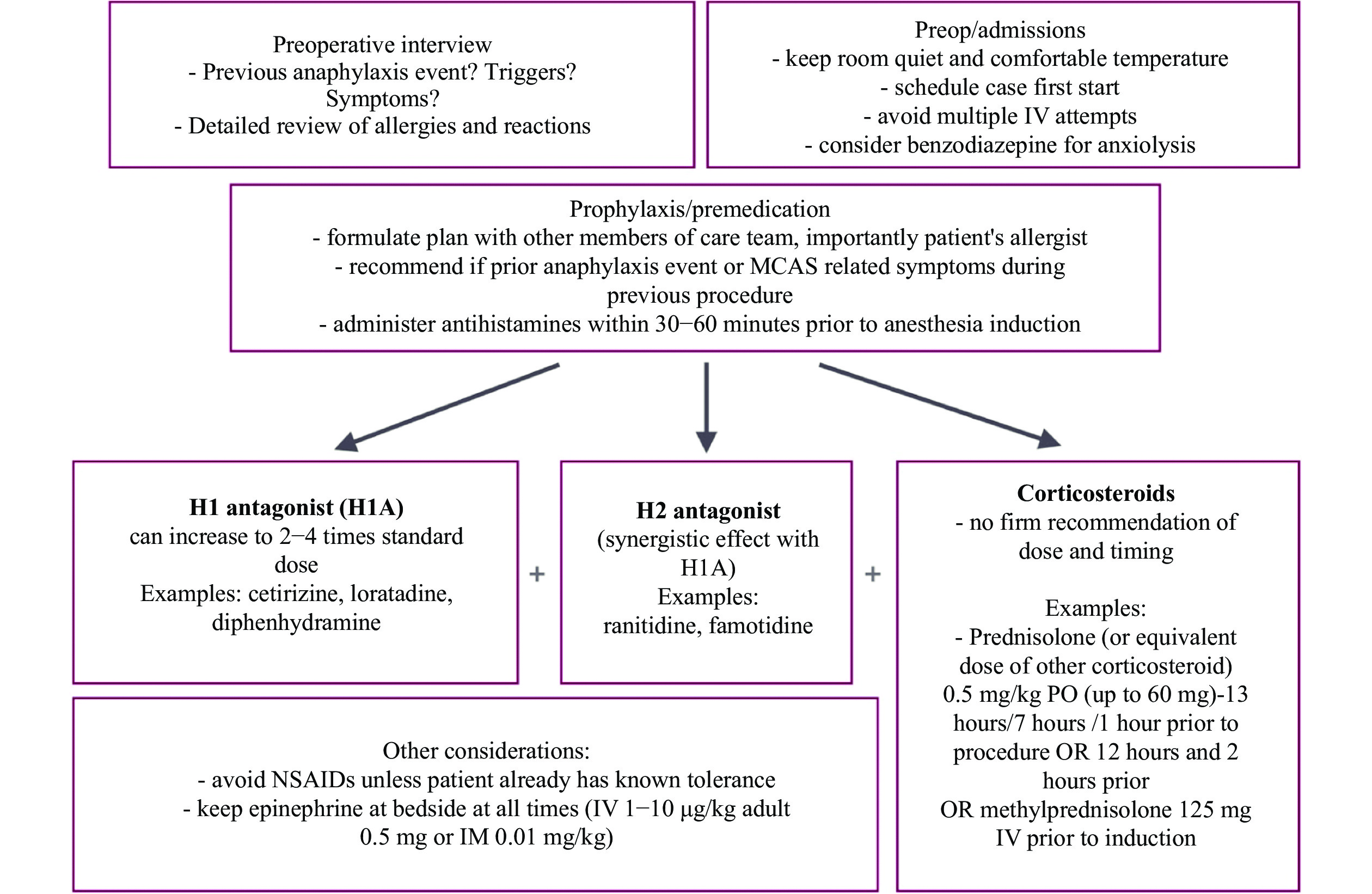
Mast cell activation syndrome (MCAS) includes a group of disorders that result in the inappropriate release of inflammatory mediators from mast cells. These mediators can affect multiple organ systems and lead to significant morbidity, and possible fatality. Although reactions, typically in response to various nonspecific stimuli, are usually mild, they may put those with MCAS at increased risk of anaphylaxis. In this case report, we present two clinical scenarios of MCAS, and identify possible factors triggering mast cell mediator release. We also define a preoperative preventive pathway, outline anesthetic considerations, and discuss the management of immediate hypersensitivity reactions in patients with MCAS. Meticulous preoperative preparation, avoidance of triggers, and development of a plan to treat possible adverse organ responses are paramount of good outcomes.
Lassa fever (LF) is a viral illness of endemic proportion amongst the population of Western Africa[1]. The causative agent of the disease in mammals is Lassa mammarenavirus (LASV), while the rodent Mastomysnatalensis is the main carrier and transmitter of the disease[2]. Lassa virus, the most virulent arenavirus, belongs to the Old World (OW) viruses[3]. Each year 300 000 to 500 000 infections are reported in West Africa, with approximately 5000 deaths due to LASV infection[4]. The LASV can spread via zoonotic and human-to-human transmission as a result of direct or indirect contact with biological fluids (urine, blood, saliva, feces) from infected hosts[5]. Fever, nausea, and hemorrhage, along with neurological, pulmonary, and gastrointestinal complications, are the major symptoms of LF which take typically 3 to 21 days to manifest[6]. The virion glycoprotein (GP) is the main regulatory element for the LASV entry inside the host. The GP1, from the SSP-GP1-GP2 trimer, attaches to α-dystroglycan (α-DG) receptor on the host cells[7]. The dystroglycan interlinks the extracellular matrix to the cytoskeleton and is predominately expressed in all types of human tissues[8]. For normal biological activities, α-DG needs to undergo post-translational glycosylation by glycosyltransferase, and this glycosylation is also a prerequisite for the recognition and binding of the LASV GP1 to α-DG[9]. The attachment of the virion glycoproteins with extracellular α-DG induces the phosphorylation in the tyrosine residues of β-dystroglycan, which eventually facilitates the transportation of the viral particle to the intracellular late endosomal compartment[10]. Once inside, GP is cleaved into stable signal peptide (SSP) and premature GP1/GP2 complex. After getting translocated in the Golgi apparatus, GP1/GP2 complex is cleaved into N terminus GP1 and C terminus GP2 subunit[11]. Afterward, the GP1 alters its conformity at a low acidic environment (~pH 5) and promotes the switching of its receptor from α-DG to lysosome-associated membrane protein 1 (LAMP1) and the consequent membrane fusion, replication, transcription, and translation of viral genome[7]. Thus, blocking the GP1 and α-DG receptor complex formation can prevent LASV entry into host cells and disrupt the entire infection process.
Experimental identification of a random viral epitope involves many expensive and time-consuming steps ranging from antibody production to antigenic region mapping on a target protein, obtaining crystals of antigen-antibody complexes, determination of their 3D structures by X-ray crystallography, etc[12]. Thus, identifying and utilizing antiviral drugs could serve as a convenient strategy over the arduous process of new vaccine development. Plant-derived naturally occurring chemical constituents have been thoroughly studied for their anti-infective activity along with many other medicinal uses (such as antioxidants, anti-inflammatory, and analgesic agents). A wide spectrum of phytochemicals have been reported to have excellent antiviral effects, amongst which, phenolics, carotenoids, terpenoids, and alkaloids show substantial activity against HSV-1, HCV, poliovirus, avian influenza virus, dengue virus, etc[13]. Tangeretin (pentamethoxyflavone) can block Lassa virus propagation by interrupting the LASV replication cycle[14]. Therefore, screening naturally occurring compounds for anti-infective activity against LASV presents as a promising strategy.
This research aimed to establish a library of compounds to identify the most drug-like candidates that can block the human α-DG receptor, and prevent the insertion and propagation of LASV inside the host cells. ADMET and quantitative structure-activity relationship (QSAR) assessment, molecular docking, and molecular dynamics simulation were performed to explore the pharmacokinetic properties of the candidate ligands, inspect the efficacy and orientation of their binding, and examine the stability of receptor-ligand complexes respectively. The goal was to screen out the lead compounds to develop and formulate targeted therapeutics to inhibit the LASV infection.
Materials and methods
Construction of the compound library
The α-DG receptor is a transmembrane protein that acts as a receptor for extracellular matrix proteins which contain laminin G domains[15]. After thoroughly searching the ChEMBL (https://www.ebi.ac.uk/chembl/) database, only one small molecule (PubChem CID: 127037754) was found with human α-DG modulatory activity[16]. With its Simplified Molecular Input Line Entry System (SMILES) notation, a pharmacokinetic profile was built for this molecule in 'Swiss ADME' (http://www.swissadme.ch/index.php). Then a thorough literature review was performed incorporating previous research, clinical data, and non-clinical observations on arenaviruses, phytochemicals (i.e., carotenoids, phytosterols, limonoids, flavonoids, isoflavonoids, isoprenoids, etc.), and bioactive compounds. Finally, 483 compounds with anti-microbial, anti-infective, and anti-viral activities (either broad-spectrum or arenavirus-specific) were shortlisted. From these compounds, 200 compounds with overlapping chemical or pharmacokinetic profiles to the aforementioned α-DG modulator (e.g., presence of benzene ring or cyclohexene, number of heavy atoms, number of H-bond acceptors, number of H-bond donors, etc.) were selected. Then the 3D conformers of these 200 compounds were retrieved from PubChem (https://pubchem.ncbi.nlm.nih.gov/), an NCBI authorized open chemistry repository, to construct the compound library for this study. The phytochemical nobiletin was selected as the 'Control' ligand for this study as it has anti-viral and anti-microbial activities and has exhibited the capacity to block LASV infection (≈22% inhibition) in in-vitro studies[17].
Screening the ligand library via ADMET and QSAR profiling for ligand validation
The reported and predicted pharmacokinetic properties of the 200 ligands were profiled. The ADMET assessment could help to predict the disposition of pharmaceutical compounds inside the body[18]. 'Swiss ADME' (http://www.swissadme.ch/index.php) and 'Molinspiration Cheminformatics' (https://www.molinspiration.com/cgi-bin/properties) were utilized to build the absorption, distribution, metabolism, elimination, and synthetic accessibility profile of these compounds. The toxicity assessment was performed on 'pkCSM' (http://biosig.unimelb.edu.au/pkcsm/prediction) and 'admetSAR 2' (http://lmmd.ecust.edu.cn/admetsar2/). All the ligands were compared to the control ligand nobiletin and ranked based on their ADMET profiles to find the top drug-like candidates. Finally, the quantitative structure-activity relationship (QSAR) assessment for the top ligands was carried out on the PASS server (http://www.pharmaexpert.ru/passonline/) for validation of anti-infective, anti-viral, or anti-microbial activity.
Optimization of components
Optimization of macromolecule
A high resolution (1.8 Å) 3D crystal structure of the chain 'A' of human α-DGn was selected from the protein data bank (PDB ID: 5LLK). Firstly, the FASTA sequence of the resolved protein was obtained and used in the SEQATOMs web interface (https://www.bioinformatics.nl/tools/seqatoms/)[19] to identify any missing residues in the crystal structure. The BLAST output from SEQATOMs indicated missing residues at the N-terminus, C-terminus, and in the middle region of the resolved structure of the protein. The FASTA sequence was then uploaded to the COACH-D server (https://yanglab.nankai.edu.cn/COACH-D/)[20] to obtain a homology model of the chain 'A' of human α-DG without any missing residues, and predict the top five protein-ligand binding pockets. Parallelly, the FASTA sequence was used in SWISS-MODEL (https://swissmodel.expasy.org/)[21], with 5LLK as the template, to obtain another homology model (with the missing residues modeled and filled only in the middle region). The 'pdb' file of this homology model was then uploaded to the COACH-D server for the prediction of top protein-ligand binding pockets. Afterward, the 3D crystal structure of α-DG (PDB ID: 5LLK) was obtained from the protein data bank[22]. The X-ray structure was optimized to present the proper size, orientation, and rotations of the protein crystal structure[23]. Here, the macromolecule was optimized using UCSF Chimera (version 1.14) (https://www.cgl.ucsf.edu/chimera/) to remove non-standard amino acids, water molecules, ligands, and ions, add missing hydrogen atoms, and perform energy minimization of the protein structure[24]. The PDB file of the optimized protein was then uploaded to the COACH-D web interface for the prediction of top protein-ligand binding pockets. Afterward, all the three outputs from the COACH-D server were compared to see if the previously missing residues were potential sites for protein-ligand interaction. None of the predicted top binding pockets from the homology models of α-DG suggested these residues to be important for binding interaction. The homology model obtained from SWISS-MODEL had the most nativelike structure and the most comprehensive binding pocket predictions. Thus, this protein model was utilized as the macromolecule after adding missing hydrogen atoms and energy minimization in UCSF Chimera. The protein structure has been shown in Fig. 1A, along with the supramolecular docking pose with a probe ligand to depict the best active site for ligand binding. Ideally, if a ligand can interact with the amino acid residues in this site, and yield binding energy of −5.6 kcal/mol, the binding can be considered effective.
Figure
1.
Illustration of all the optimized ligands and receptor macromolecule simultaneously.
A: α-dystroglycan with its active site for complexing with the ligands are presented inside the grid box, while the probe ligand suggests the main conformation of the pocket. B–H: Nobiletin (B) is the control ligand, and the candidate ligands have been depicted as reticuline (C), kirenol (D), chrysin (E), 3-caffeoylshikimic acid (F), gigantamide A (G), and dasyclamide (H).
3D structures of the selected ligands were obtained in 'sdf' format from PubChem[25]. Energy minimization was done to reduce the accumulative charge on ligands to zero based on the Gasteiger method[26] in UCSF Chimera (version 1.14). After optimization, the selected ligands were transformed into a 'mol2' file for further assessment and molecular docking. All optimized structures of the ligands have been shown in Fig. 1B–H.
Operation of molecular docking
Molecular docking was conducted with the optimized protein and ligands using PyRx 0.8 docking software[27]. The grid box area was set at, X=48.5488 Å, Y=53.1510 Å and Z=71.5823 Å where the center of the grid box was X:Y:Z=26.0331:7.5869:26.6648 respectively, to ensure that all the binding site amino acid residues predicted by COACH-D were covered. Here, the selected macromolecule and ligands were converted into 'pdbqt' format. After completing the docking, the binding affinity of the ligands was calculated, and root mean square deviations (RMSD) were saved in the 'csv' file for individual protein-ligand complexes. Fig. 2 illustrates the binding poses for the control and top ligands.
Figure
2.
The prediction of the diversified docking poses of the ligands depending on the active site region of the protein.
The control ligand nobiletin (A) is complexed with α-dystroglycan, while the test ligands complexed with the receptor protein are reticuline (B), kirenol (C), chrysin (D), 3-caffeoylshikimic acid (E), gigantamide A (F), and dasyclamide (G) respectively.
The initial visualization of receptor-ligand interactions was conducted via Discovery Studio Visualizer (https://accelrys-discovery-studiovisualizer.software.informer.com/3.0/). Afterward, PyMOL (version 2.4.1) (https://pymol.org/) was used to further analyze and visualize these interactions. The protein-ligand complexes were saved as 'pdb' files for further analysis. Finally, the saved files were analyzed in Ligplot+ (version 2.2) (https://www.ebi.ac.uk/thornton-srv/software/LigPlus/) to mark the potential hydrogen bond interactions and scopes for non-covalent bond formation[28]. Retrospective docking validation was performed using decoy ligands. The decoy ligands for the control and the top six candidates were obtained from DUD-E server (http://dude.docking.org/)[29]. Though the decoys physically resembled these compounds, they were unlikely to bind due to topological dissimilarities. The binding pocket in the protein was redefined based on the Ligplot+ (version 2.2) output and the docking simulations were repeated with the decoys to eliminate any false-positive result. The relative binding affinity of the control and candidate ligands against the α-DG receptor was assessed using the 'Molecular Mechanics Generalized Born Surface Area' (MMGBSA) method in Maestro-Desmond (https://www.schrodinger.com/products/maestro).
Molecular dynamics simulation
Firstly, molecular dynamics simulation (MDS) of the ligand-free protein was run on CABS-flex 2.0 (http://biocomp.chem.uw.edu.pl/CABSflex2) for 10 nanoseconds to observe its natural changes in structural orientation[30]. Afterward, MDS for individual protein-ligand complexes were run on the LARMD molecular dynamics simulator (http://chemyang.ccnu.edu.cn/ccb/server/LARMD/index.php)[31] up to 1.1 nanoseconds for an initial analysis of principal component analysis (PCA), RMSD, root mean square fluctuation (RMSF), solvent accessible surface area (SASA) and Debye-Waller factor for thermostability (B-Factor). Finally, MDS was run on GROMACS molecular dynamics package (version 5.1.2) with the protein-ligand complexes to analyze RMSD, RMSF, SASA, B-Factor, and Polar Surface Area (PSA) for a runtime of 100 nanoseconds. The box size was set to 10:10:10, along with neutralizing ions (Na+) as required (4 Na+ ions for chrysin and 3-caffeoylshikimic acid, 3 Na+ for nobiletin, reticuline, kirenol, gigantamide A, and dasyclamide). Chrysin had the highest number of system atoms (35677) and the rest followed as 3-caffeoylshikimic acid (34660), kirenol (34654), reticuline (34649), nobiletin (34644), dasyclamide (34643), and gigantamide A (34638). The probe radius was set to 1.4 Å for SASA and molecular surface area (MolSA) analysis.
Statistical analysis and graphical representation
The data generated from molecular docking and MDS for PSA, RMSD, RMSF, SASA, MolSA, and Radius of gyration (Rg) were statistically analyzed using R programming (version R-4.0.2)[32–33] and GraphPad Prism (version 8.0.1; GraphPad Software, USA)[34–35]. The protein-ligand complexes were refined and graphically visualized using the aforementioned software packages.
Results
ADMET and QSAR profiling indicated 6 compounds with the most drug-like potential
The initially established library of 200 compounds was screened based on ADMET and QSAR profiles, revealing 6 candidates that possessed the most drug-like potential. These compounds were expected to reach the site of action and yield desirable biological activity. The pharmacokinetic properties of nobiletin and the 6 promising candidates have been listed in Table 1. None of these compounds showed any violation of Lipinski's rules. The six candidates exhibited good excretion rates from the body after metabolism, while expressing maximum tolerated doses in the range of −0.47 to 0.666 log mg/(kg·day). All were predicted to be mostly absorbed (>90%) from the intestine, except 3-caffeoylshikimic acid (45.512%) (PubChem CID: 10131826). Only reticuline (PubChem CID: 439653) and chrysin (PubChem CID: 5281607) were predicted to cross the blood-brain barrier. None of the top six molecules demonstrated any potential for hepatoxicity or AMES toxicity, and the LD50s were in the range of 2.068 to 2.556 mg/kg. The top 6 candidate molecules were ordered based on their synthetic accessibility score as follows: dasyclamide (2.66), chrysin (2.93), reticuline (3.07), gigantamide A (3.47), 3-caffeoylshikimic acid (4.20), and kirenol (5.42). All the top six molecules exhibited bioactivity either as anti-infective, anti-viral, or anti-microbial agents (Table 2).
Table
1.
Pharmacokinetics profiling of ADMET and QSAR for ligand validation
Ligands
MW
H-Ac
H-Do
Log P
NRB
IA (%)
BBB
TC
LD50
HT
AT
MTD
NLV
DL
Nobiletin
402.39
8
0
3.02
7
98.921
No
0.789
2.459
No
No
0.443
0
Yes
Reticuline
329.39
5
2
2.6
4
91.276
Yes
1.04
2.296
No
No
0.232
0
Yes
Kirenol
338.48
4
4
2.32
3
92.674
No
0.849
2.109
No
No
0.666
0
Yes
Chrysin
254.24
4
2
2.55
1
93.761
Yes
0.405
2.289
No
No
0.016
0
Yes
3-Caffeoylshikimic acid
336.29
7
5
0.21
4
45.512
No
0.406
2.068
No
No
–0.47
0
Yes
Gigantamide A
314.38
3
2
1.82
7
94.153
No
0.539
2.341
No
No
–0.496
0
Yes
Dasyclamide
316.39
3
3
2.03
9
94.535
No
0.691
2.556
No
No
0.057
0
Yes
ADMET: absorption, distribution, metabolism, excretion, and toxicity; QSAR: quantitative structure-activity relationship; MW: molecular weight; H-Ac: No. of hydrogen bond acceptor; H-Do: No. of hydrogen bond donor; LogP: predicted octanol/water partition coefficient; NRB: No. of rotatable bonds; IA: intestinal absorption; TC: total clearance, log mL/(min·kg); LD50: oral rat acute toxicity, mg/kg; BBB: blood brain barrier; HT: hepatotoxicity; AT: AMES toxicity; MTD: maximum tolerated dose for a human, log mg/(kg·day); NLV: No. of Lipinski's rule violations; DL: drug-likeness.
Table
2.
QSAR based bioactivity prediction for ligand validation
Compounds
Prediction of activity spectra for substances (Pa = 0.3 to 0.7)
Anti-infective
Anti-viral
Anti-microbial
Nobiletin
×
√
√
Reticuline
×
×
√
Kirenol
×
√
√
Chrysin
√
√
√
3-Caffeoylshikimic acid
√
√
√
Gigantamide A
×
√
×
Dasyclamide
×
√
√
QSAR: quantitative structure-activity relationship; Pa: prediction of activity score as per PASS server. Pa score of 0.3 to 0.7 signifies moderate activity. ×: Pa score less than 0.3; √: Pa score in the range 0.3 to 0.7
Before energy minimization in UCSF Chimera, the energy level of the protein was –52 407.7 kJ/mol. After energy minimization, the energy level of the protein was –113 291.2 kJ/mol. The energy levels before and after the energy minimization step were determined using YASARA (https://www.yasara.org)[36]. To unveil the most suitable ligands among the six selected compounds, the virtual screening tool PyRx 0.8 was employed to generate docking scores of the best fitting protein-ligand complex. In the PyRx system, the best docking pose between protein and ligand was determined by the highest binding affinity score. The control ligand nobiletin (PubChem CID: 72344) exhibited binding energy of −7.2 kcal/mol within the grid box area X=48.5488 Å, Y=53.1510 Å and Y=71.5823 Å, where the center of the grid box was X:Y:Z=26.0331:7.5869:26.6648, respectively. The ligands chrysin and reticuline showed the highest binding energies (−7.9 kcal/mol and −7.7 kcal/mol, respectively). On the contrary, dasyclamide (PubChem CID: 10980024) and gigantamide A (PubChem CID: 23655945) exhibited the lowest binding energy values (−5.7 kcal/mol and −5.8 kcal/mol, respectively). The remaining two compounds: 3-caffeoylshikimic acid and kirenol (PubChem CID: 15736732) showed binding energies of −7.0 kcal/mol and −6.9 kcal/mol respectively with the α-DG receptor.
After post-docking analysis of the control ligand using Ligplot+ (version 2.2), the complex formed by nobiletin with α-DG was found to be stabilized by one hydrogen bond interaction with Arg76 (2.86) residue and four hydrophobic bond interactions with Pro304, Lys302, Arg234, Ser259 residues (Fig. 3). On the other hand, three hydrogen bonds (with Arg76 [2.93 Å], Asn224 [3.20 Å], and Lys261 [3.00 Å]) and five hydrophobic bond interactions (with Lys302, Ser259, Val190, Lys226, and Trp260) were formed by reticuline against the receptor (Fig. 4A). Kirenol stabilized its complex with the α-DG receptor via four hydrogen bonds (with Glu159 [3.31 Å], Ser162 [2.96 Å], Arg76 [2.96 Å], and Ser259 [2.92 Å]) and four hydrophobic interactions (with Lys226, Lys261, Val188, and Val190) (Fig. 4B). During the inspection of the chrysin docked α-DG complex (Fig. 4C), it was observed that two hydrogen bonds were formed with Arg76 (3.34 Å) and Asn224 (2.96 Å) residues of the α-DG receptor, along with four non-conventional bonds (with Lys261, Lys302, Ser259, and Trp260). After docking of 3-caffeoylshikimic acid on the α-DG receptor, multiple hydrogen bonds (with Lys226 [3.11 Å], Thr192 [3.18 Å], Ser259 [2.84 Å and 2.84 Å], Arg76 [2.82 Å], Asn224 [3.00 Å and 3.11 Å]) and five hydrophobic bond interactions (with Leu257, Met225, Lys261, Val190, and Lys302) were identified (Fig. 4D). The complex of gigantamide A with α-dystroglycan was formed with one hydrogen bond (with Lys302 [3.25 Å]) and four hydrophobic bond interactions (with Val188, Lys226, Val190, and Ser259, Fig. 4E). Furthermore, the complex of dasyclamide with α-DG (Fig. 4F) was stabilized by one hydrogen bond (with Glu159 [3.02Å] residue) and five hydrophobic interactions (with Lys302, Lys226, Val190, Thr192 and Ser259 residues).
Figure
3.
The complex formed between the control ligand nobiletin and α-dystroglycan.
The qualitative positioning of the ligand embedded inside the alpha helix and beta sheets (as shown in the left); along with the quantitative measurements referring to the hydrophobic interactions of the amino acid residues present (as shown on the right), and hydrogen bonding distances (2.85Å with Arg76).
Figure
4.
The hydrophobic interactions and hydrogen bonds formation between each of the ligands and the amino acid residues of the receptor at the post docking stage.
Interactions between reticuline and α-dystroglycan (A), kirenol and α-dystroglycan (B), chrysin and α-dystroglycan (C), 3-caffeoylshikimic acid and α-dystroglycan (D), gigantamide A and α-dystroglycan (E), and dasyclamide and α-dystroglycan (F). The green dotted lines with the distances in Å represent hydrogen bonds formation between the ligands and the amino acid residues, when the red dotted lines indicate the potential hydrophobic interactions between the ligands and amino acid residues of α-dystroglycan.
Repeated docking simulations with the decoy ligands (for the control and candidate ligands) revealed that none of the observed protein-ligand interactions were false positive outcomes. Then the binding-free energy of the control and candidate ligands against the α-DG receptor was assessed using the MMGBSA method in Maestro-Desmond. All the MMGBSA binding scores (kcal/mol) obtained for the ligands were high negative values. The MMGBSA scores have been summarized (Table 4).
Table
4.
Binding free energy assessment using MM/GBSA
Protein
Ligands
MM/GBSA binding score (kcal/mol)
α-DG
3-caffeoylshikimic acid
−45.89
α-DG
Kirenol
−29.06
α-DG
Chrysin
−24.72
α-DG
Nobiletin
−42.89
α-DG
Dasyclamide
−34.74
α-DG
Gigantamide A
−38.44
α-DG
Reticuline
−30.84
α-DG: α-dystroglycan; MM/GBSA: molecular mechanics/generalized born surface area.
The maximum value of RMSD was 3.25 Å with the control ligand nobiletin in complex with the optimized macromolecule. The RMSD fluctuations of the ligands have been shown in Fig. 5A–H. Whereas the ligands dasyclamide and kirenol exhibited higher RMSD values of 3.56 Å and 3.33 Å respectively. The other compounds, such as gigantamide A, reticuline, chrysin, and 3-caffeoylshikimic acid showed high RMSD values of 3.19 Å. 3.07 Å, 2.97 Å, and 2.78 Å respectively. The lowest RMSD values 0.99 Å, 0.98 Å, and 0.89 Å were exhibited by the compounds gigantamide A, chrysin, and 3-caffeoylshikimic acid. The RMSF profiles of all the protein-ligand complexes were obtained for 225 protein residues with the most notable fluctuations (Fig. 6A–H). The control ligand exhibited RMSF values ranging from 0.378 Å up to 7.49 Å. The ligands kirenol and dasyclamide have shown higher fluctuations compared to nobiletin (RMSF values ranging 0.4 to 10.63 Å and 0.41 to 8.62 Å respectively). The ligands 3-caffeoylshikimic acid, gigantamide A, chrysin, and reticuline had lower fluctuations compared to the control.
Figure
5.
The RMSD values of each of the individual ligand-receptor complexes obtained from the molecular dynamics simulation (100 nanoseconds).
The RMSD fluctuations with a diversified range of thresholds, making each complex unique from the others. Here, the fluctuations in the receptor and ligand structures have been shown for nobiletin and α-dystroglycan (A), reticuline and α-dystroglycan (B), kirenol and α-dystroglycan (C), chrysin and α-dystroglycan (D), 3-caffeoylshikimic acid and α-dystroglycan (E), gigantamide A and α-dystroglycan (F), and dasyclamide and α-dystroglycan (G). RMSD: root-mean-square deviation.
Figure
6.
The RMSF values resulted from the molecular dynamics simulation (100 nanoseconds).
A–G: The RMSF fluctuations for the ligands nobiletin (A), reticuline (B), kirenol (C), chrysin (D), 3-caffeoylshikimic acid (E), gigantamide A (F), and dasyclamide (G) in complex with α-dystroglycan. H: The fluctuation range of alpha carbon (Cα) atoms involved for each ligand with respect to their RMSF thresholds have been provided as whiskers. Here, the Cα fluctuations have been depicted for nobiletin (a), reticuline (b), kirenol (c), chrysin (d), 3-caffeoylshikimic acid (e), gigantamide A (f), and dasyclamide (g).
The control ligand along with the rest six selected ligands: reticuline, kirenol, chrysin, 3-caffeoylshikimic acid, gigantamide, and dasylclamide, all yielded the polar energy at 4620.04 and apolar energy at 7575.65 in terms of the total area to energy ratio, with no unknown area (Table 5). Reticuline, gigantamide A, and dasyclamide presented higher SASA (55.30 to 381.21 Å2, 62.49 to 369.35 Å2, and 39 to 308.08 Å2 respectively) values when compared to the nobiletin. kirenol (35.23 to 220.724 Å2), 3-caffeoylshikimic acid (44.8 to 182.89 Å2), chrysin (32.6 to 180.92 Å2) exhibited SASA values in the lowest ranges (Fig. 7A).
Table
5.
Solvent accessible surface area referring the area to energy ratio over the entire dynamic simulation process (100 nanoseconds) with polar and apolar regions precisely
Macromolecule
Ligands
WPR (Å)
GIC
TNR
Total area/energy (Ų/[kcal·mol])
Polar
Apolar
Unknown
α-DG
Nobiletin
1.4
No
244
4620.04
7575.65
0.00
α-DG
Reticuline
1.4
No
244
4620.04
7575.65
0.00
α-DG
Kirenol
1.4
No
244
4620.04
7575.65
0.00
α-DG
Chrysin
1.4
No
244
4620.04
7575.65
0.00
α-DG
3-caffeoylshikimic acid
1.4
No
244
4620.04
7575.65
0.00
α-DG
Gigantamide A
1.4
No
244
4620.04
7575.65
0.00
α-DG
Dasyclamide
1.4
No
244
4620.04
7575.65
0.00
WPR: water probe radius; GIC: gradient in calculation; TNR: total No. of residues.
Figure
7.
The four major parameters with significant scores emerging from the molecular dynamics simulation.
The illustration of the molecular dynamics simulation output for the parameters of solvent accessible surface area (SASA) (A), molecular surface area (MolSA) (B), radius of gyration (Rg) (C), and polar surface area (PSA) (D) for a runtime of 100 nanoseconds. In each sections, the fluctuations observed have been depicted for nobiletin (a), reticuline (b), kirenol (c), chrysin (d), 3-caffeoylshikimic acid (e), gigantamide A (f), and dasyclamide (g).
A probe radius of 1.4 Å was taken to calculate the MolSA, as that radius is equivalent to the van der Waals surface area of a water molecule. The MolSA values fluctuated over most of the simulation time (100 nanoseconds) at varying ranges. dasyclamide and gigantamide A complexes showed the largest MolSAs (353.18 Å2 and 338.08 Å2) with the widest fluctuations. The other four protein-ligand complexes with reticuline, kirenol, chrysin, and 3-caffeoylshikimic acid showed fewer fluctuations in MolSA during the simulations (Fig. 7B). For these ligands, the highest MolSA was observed at 318.01 Å2 (reticuline), whereas the least MolSA was observed to be 226.42 Å2 (chrysin).
With the control ligand nobiletin, the Rg was observed in the range of 4.29–4.51 Å. Considering the lowest values observed for Rg, the ligands reticuline (3.71 Å), kirenol (3.46 Å), gigantamide A (3.38 Å), dasyclamide (3.34 Å), and chrysin (3.3 Å) showed lower Rg values than the control ligand. Considering the highest values observed, only reticuline (4.38 Å), kirenol (3.9 Å), and chrysin (3.45 Å) exhibited lower Rg values than the control ligand. A high degree of variability in the Rg was observed for the ligands gigantamide A and dasyclamide (Fig. 7C).
Throughout the MDS, the control ligand displayed an effective polar surface area of 68.28 Å2 to 109.42 Å2. The selected ligands were ranked depending on the highest PSA values exhibited by them during the simulation runs as follows: 3-caffeoylshikimic acid > kirenol > dasyclamide > chrysin > gigantamide A > reticuline (323.06 Å2, 184.75 Å2, 154.12 Å2, 139.86 Å2, 135.86 Å2, and 126.14 Å2, respectively). Conversely, based on the lowest values observed for PSA, the ligands can be ranked as dasyclamide < gigantamide A < reticuline < chrysin < kirenol < 3-caffeoylshikimic acid (83.44 Å2, 85.91 Å2, 97.21 Å2, 125.22 Å2, 157.07 Å2, and 292.5 Å2, respectively). Either way, all the ligands showed higher PSA values than the control ligand nobiletin (Fig. 7D).
The occupancy of the ligand-protein hydrogen bonds (H-bonds) was a major parameter to be analyzed from the molecular dynamics simulation (MDS) part. In this study, the frequency of the occupancy of hydrogen bonds was rated in '%', where nobiletin (control), reticuline, kirenol, chrysin, 3-caffeoylshikimic acid, gigantamide A and dasyclamide, in complexing with the α-DG receptor showed- 22.078%, 88.71%, 53.55%, 92.11%, 37.36%, 65.93%, and 23.78%, respectively. The estimation of the occupancy (%) was calculated from 1001 frames of MDS for each ligand-protein complex individually. In that case, α-DG with the reticuline, chrysin, 3-caffeoylshikimic acid, and gigantamide A showed strong H-bonds formations between their interfaces.
Discussion
Currently the only effective therapy for LF is intravenous (Ⅳ) infusion of Ribavirin, though it is not an approved indication of this medication[1]. Ⅳ administration of Ribavirin has been observed to be more effective, the earlier it is used in the course of therapy (i.e., within 6 days of diagnosis)[37]. The symptoms of LF take 1 to 3 weeks to manifest after the first exposure, and the cases with mild symptoms often remain undiagnosed[1]. Thus, there remains an unmet need for a prophylactic treatment option for suspected cases before a confirmed diagnosis of LF. Identification of compounds that can block LASV entry into host cells can be very useful to address this unmet need.
Ribavirin has been shown to inhibit the activity of the host inosine monophosphate dehydrogenase (IMPDH) that results in the disruption of critical viral replication steps[38–40]. An additional ribavirin metabolite, ribavirin triphosphate, has been reported to exhibit mutagenic activity that can inhibit virus replication[41]. Several other drugs like stampidine (retroviral reverse transcriptase inhibitor), arbidol (inhibits viral fusion), 5-ethynyl-1-b-D-ribofuranosylimidazole-4-carboxamide (EICAR) (inhibits IMPDH), mycophenolic acid (inhibits IMPDH), and isavuconazole (targets GP2 and inhibits cell to cell viral fusion) have been reported to be effective against LASV infection in animal and in-vitro studies[37,42]. Recently, several compounds have been reported to inhibit the low-pH-induced membrane fusion step in LASV infection in in-vitro studies, such as casticin[43], lacidipine, and phenothrin[44]. Considering all the aforementioned drugs and strategies against LASV infection, the discovery of drugs that can block LASV entry into host cells is of utmost importance in the absence of any approved vaccine. Isavuconazole, an anti-fungal drug, has been reported to inhibit LASV entry by targeting the SSP-GP2 subunit[45]. Recently, a potent small-molecule LHF-535 targeting the viral envelop glycoprotein has been developed and proven to be effective in preventing LASV entry in animal models of the guinea pig and cynomolgus macaque[46]. But its developers have also reported the risk of drug resistance by V434I substitution in the transmembrane domain of the envelope glycoprotein GP2 subunit[47]. So, finding an alternative strategy other than targeting the viral glycoprotein is also required to block the LASV entry into host cells. Until now, no natural or synthetic compound has been reported to inhibit LASV entry into host cells by blocking the host α-DG receptor. Thus, this study focused on finding such compounds with potential for prophylactic use against LF.
The molecular docking helped to identify and analyze the probable protein-ligand interactions for the control and six selected ligands. The control ligand exhibited good binding affinity to the α-DG receptor (−7.2 kcal/mol) while forming one hydrogen bond with the Arg76 residue of the protein and four hydrophobic interactions with the residues Pro304, Lys302, Arg234, and Ser259. Compared to the control ligand, the two ligands with the best binding affinity were chrysin and reticuline (−7.9 kcal/mol and −7.27 kcal/mol respectively). Unlike the control ligand, these two ligands formed multiple hydrogen bonds, including the common interaction with the Arg76 residue (Table 3). Other common features of the binding of these three ligands were multiple hydrophobic interactions with the residues Lys302 and Ser259. Though dasyclamide managed to interact with the residue Arg76 via one hydrogen bond formation, it lacked any hydrophobic interaction with Lys302 and Ser259 residues and showed a comparatively lesser binding affinity for the target protein. After MMGBSA assessment, the control ligand and the top six candidates returned with high negative values for biding-free energy, suggesting good binding affinity with the α-DG receptor.
Table
3.
Analysis of the binding affinities of the candidate ligands with α-dystroglycan
MDS enables the investigation of the spatial arrangement of protein residues and ligands within the protein-ligand complexes and the relevant comparisons to reveal the most stable binding interactions[48]. GROMACS molecular dynamics simulator has been utilized for MDS as it can utilize both GPU and CPU for heterogeneous parallelization of computation and subsequent accelerated high-quality output[49]. MDS was run for every protein-ligand complex for 100 nanoseconds to analyze the RMSD, RMSF, PSA, Rg, MolSA, and SASA. Ideally, the ligands which result in the RMSD value ≤ 2 Å for the protein backbone in the protein-ligand complexes are considered to have achieved good binding pose with high accuracy of molecular docking simulation[50].
RMSD is used to assess the average change in displacement of a selection of atoms for a particular frame compared to a reference backbone frame. It is calculated for all frames in the trajectory. After comparing the RMSD values for the selected ligands with that of the control ligand, the binding poses for 3-caffeoylshikimic acid, chrysin, reticuline, and gigantamide A were found to be the ones nearest to the true binding poses, setting the threshold for the highest RMSD value at 3.19 Å (gigantamide A) and the lowest RMSD value at 0.89 Å (3-caffeoylshikimic acid).
To investigate how ligand binding affects the backbone atoms of the macromolecule, the RMSF profile was compiled for all of the protein-ligand complexes. The higher RMSF values pointed towards greater destabilization of the key regions in the protein structure due to increased flexibility[51]. RMSF data represent the flexibility and strength of enzyme-substrate complexes (Fig. 6). Similar to the pattern observed with RMSD, the ligands kirenol and dasyclamide showed higher RMSF values when compared to the control ligand. Thus, these ligands can increase the flexibility of the protein backbone atoms and consequently result in decreased stability of the protein-ligand complex. The ligands 3-caffeoylshikimic acid, gigantamide A, chrysin, and reticuline showed RMSF values in the considerable range with the highest threshold at 7.21 Å and the lowest value being 0.41 Å. Thus, these ligands may be able to form more stable complexes with α-DG than the control ligand.
The SASA values were analyzed for better comprehension of the effective interaction in the receptor-ligand complexes. This interpretation consisted of the interaction between the surface of the macromolecule-ligand complex and the water molecules surrounding the complex. The values were based on the total area to energy ratio. The compounds for which the highest SASA values were observed, their protein-ligand complexes were deemed to be the most solvent accessible, and thus unstable. Whereas, the complexes with the lowest values of SASA were considered superior in terms of stability (Fig. 7A). A similar pattern was observed for the compounds dasyclamide and gigantamide A in terms of Rg, MolSA, and SASA (Fig. 7A–C), all yielding protein-ligand complexes with lower stability than the control ligand. This is understandable as high values of SASA can be attributed to both high molecular surface area[52] and high radius of gyration values[53]. On the other hand, the ligands kirenol and chrysin present with lower Rg, MolSA, and SASA readings and thus, are deemed to produce more stable protein-ligand complexes compared to the control ligand (Fig. 7A–C).
PSA is one of the most important parameters to consider for assessing the capacity of compounds to cross the blood-brain barrier[54]. The linear transformation for this structure-activity relationship usually presents as a 'rump' function. The ideal PSA value 'X' for the compounds capable of crossing the blood-brain barrier lies within the range 40 Å2 < X ≤ 90 Å2, which is preceded and extended by two undesirable ranges: X ≤ 20 Å2 and X > 120 Å2 respectively[55]. Both of the highest and lowest PSA values for the control ligand nobiletin were within the desirable range. On the contrary, both of the highest and lowest PSA values for the ligands 3-caffeoylshikimic acid, kirenol, and chrysin were in undesirable ranges (292.5 Å2 to 323.06 Å2, 157.07 Å2 to 184.754 Å2, and 125.22 Å2 to 139.86 Å2 respectively). On the other hand, the lowest PSA values for the ligands dasyclamide and gigantamide A and reticuline fell within or nearest to the desirable PSA range, with their highest observed PSA values in the undesirable region. Thus, from amongst the six selected ligands, only the latter three might be able to penetrate the blood-brain barrier (Fig. 7D).
Limitations
Although the crystal structure of α-DG (5LLK) had a very high resolution (1.8 Å), there were missing residues in the resolved structure at the visible N-terminus, visible C-terminus, and in the middle region of the protein. The missing residues in the middle region were filled by homology modeling in SWISS-MODEL (with 5LLK as a template). But the gaps at the visible N and C terminus were not filled in as they were not predicted to be important for ligand binding by the COACH-D server. Thus, any topological errors in the MDS output arising from these gaps could not be addressed. Currently, there are no known small molecules that bind to the α-DG receptor. Thus, the conventional control experiments or control selection processes could not be followed here. The ligand nobiletin was selected as a 'control' ligand because it has previously exhibited limited capacity to retard the influx of LASV in cells, and good binding affinity towards α-DG in docking simulations. Though MDS in GROMACS was run for a standard 100 nanoseconds, a higher simulation time could have been more informative.
Conclusions
This insilico study incorporated construction of a library of 200 molecules and further screening based on ADMET and QSAR profiles to find the top drug-like candidates. Then molecular docking simulations with these candidates against the optimized target protein α-DG, and molecular dynamics simulation of the complexes were carried out to assess the efficacy and orientation of protein-ligand binding, and to obtain the RMSD, RMSF, PSA, radius of gyration, MolSA, and SASA values. Additionally, the assessment of protein-ligand interactions in LigPlot+ (version 2.2) led to the revelation of important amino acid residues for successful and stable binding (interactions with Arg76, Asn224, Lys302, and Ser259 residues of α-DG). The rigorous analysis of the MDS data guided the ranking of the selected 6 ligands in terms of least deviation of the protein backbone, least fluctuation of the protein alpha carbons, least flexibility of the protein, least molecular and solvent accessible surface area on the protein-ligand complexes, following the binding of the ligands to the protein. Amongst the selected six ligands, chrysin, reticuline, and 3-caffeoylshikimic acid were the top three ligands predicted to form the most stable interactions with the α-DG receptor. The findings have three major connotations. Firstly, the selected compounds, especially the top three compounds, can be further investigated in both in-vitro and in-vivo studies to determine if they can successfully suppress the influx of LASV in host cells and establish their pharmacodynamic profiles for potential therapeutic use. Secondly, based on the findings from this study, an artificial intelligence (AI) assisted virtual screening can be initiated to find new 'hits' by sampling a relevant, but comparatively bigger chemical space. Finally, ligand-based drug design (LBDD) or structure-based drug design (SBDD) ventures can be started to develop better molecules to block the α-DG receptor in humans and protect against LASV infection and the resultant LF.
Acknowledgments
The authors must extend their respect and satisfaction to RPG Organization (Govt. Registration ID: 05-060-06021) for ensuring all technical supports in cordial collaboration as part of excellent organizational technology transfer and faculty exchange under the Project 'Category F3 (ID. #08-2021/22)'.
Acknowledgments
This work was supported in part by the Department of Anesthesiology and Pain Medicine of University of California Davis Health and NIH grant UL1 TR000002 to the University of California Davis Health.
Weiler CR, Austen KF, Akin C, et al. AAAAI Mast Cell Disorders Committee Work Group Report: mast cell activation syndrome (MCAS) diagnosis and management[J]. J Allergy Clin Immunol, 2019, 144(4): 883–896. doi: 10.1016/j.jaci.2019.08.023
[2]
Valent P, Akin C, Arock M, et al. Definitions, criteria and global classification of mast cell disorders with special reference to mast cell activation syndromes: a consensus proposal[J]. Int Arch Allergy Immunol, 2012, 157(3): 215–225. doi: 10.1159/000328760
[3]
Valent P, Akin C, Nedoszytko B, et al. Diagnosis, classification and management of mast cell activation syndromes (MCAS) in the era of personalized medicine[J]. Int J Mol Sci, 2020, 21(23): 9030. doi: 10.3390/ijms21239030
[4]
Petra AI, Panagiotidou S, Stewart JM, et al. Spectrum of mast cell activation disorders[J]. Expert Rev Clin Immunol, 2014, 10(6): 729–739. doi: 10.1586/1744666X.2014.906302
[5]
Di Leo E, Donne PD, Calogiuri GF, et al. Focus on the agents most frequently responsible for perioperative anaphylaxis[J]. Clin Mol Allergy, 2018, 16: 16. doi: 10.1186/s12948-018-0094-7
[6]
Bonadonna P, Pagani M, Aberer W, et al. Drug hypersensitivity in clonal mast cell disorders: ENDA/EAACI position paper[J]. Allergy, 2015, 70(7): 755–763. doi: 10.1111/all.12617
[7]
Blunk JA, Schmelz M, Zeck S, et al. Opioid-induced mast cell activation and vascular responses is not mediated by mu-opioid receptors: an in vivo microdialysis study in human skin[J]. Anesth Analg, 2004, 98(2): 364–370. doi: 10.1213/01.ANE.0000097168.32472.0D
[8]
Matito A, Morgado JM, Sánchez-López P, et al. Management of anesthesia in adult and pediatric mastocytosis: a study of the spanish network on mastocytosis (REMA) based on 726 anesthetic procedures[J]. Int Arch Allergy Immunol, 2015, 167(1): 47–56. doi: 10.1159/000436969
[9]
By the 2019 American Geriatrics Society Beers Criteria® Update Expert Panel. American geriatrics society 2019 updated AGS beers criteria® for potentially inappropriate medication use in older adults[J]. J Am Geriatr Soc, 2019, 67(4): 674–694. doi: 10.1111/jgs.15767
[10]
Hermans MAW, Arends NJT, van Wijk RG, et al. Management around invasive procedures in mastocytosis: an update[J]. Ann Allergy Asthma Immunol, 2017, 119(4): 304–309. doi: 10.1016/j.anai.2017.07.022
Antonelli R, Forconi V, Molesti E, et al. A validated and standardized pseudotyped microneutralization assay as a safe and powerful tool to measure LASSA virus neutralising antibodies for vaccine development and comparison. F1000Res, 2024, 13: 534.
DOI:10.12688/f1000research.149578.2
2.
Nahar N, Nazmul Hasan Zilani M, Biswas P, et al. Profiling of secondary metabolite and evaluation of anti-diabetic potency of Crotalaria quinquefolia (L): In-vitro, in-vivo, and in-silico approaches. Saudi Pharm J, 2024, 32(1): 101887.
DOI:10.1016/j.jsps.2023.101887
3.
Nur Kabidul Azam M, Biswas P, Mohaimenul Islam Tareq M, et al. Identification of antidiabetic inhibitors from Allophylus villosus and Mycetia sinensis by targeting α-glucosidase and PPAR-γ: In-vitro, in-vivo, and computational evidence. Saudi Pharm J, 2024, 32(1): 101884.
DOI:10.1016/j.jsps.2023.101884
4.
Rahman MH, Al Azad S, Uddin MF, et al. WGS-based screening of the co-chaperone protein DjlA-induced curved DNA binding protein A (CbpA) from a new multidrug-resistant zoonotic mastitis-causing Klebsiella pneumoniae strain: a novel molecular target of selective flavonoids. Mol Divers, 2023.
DOI:10.1007/s11030-023-10731-6. Online ahead of print
5.
Akash S, Bibi S, Biswas P, et al. Revolutionizing anti-cancer drug discovery against breast cancer and lung cancer by modification of natural genistein: an advanced computational and drug design approach. Front Oncol, 2023, 13: 1228865.
DOI:10.3389/fonc.2023.1228865
6.
Sharif MA, Khan AM, Salekeen R, et al. Phyllanthus emblica (Amla) methanolic extract regulates multiple checkpoints in 15-lipoxygenase mediated inflammopathies: Computational simulation and in vitro evidence. Saudi Pharm J, 2023, 31(8): 101681.
DOI:10.1016/j.jsps.2023.06.014
7.
Biswas P, Bibi S, Yousafi Q, et al. Study of MDM2 as Prognostic Biomarker in Brain-LGG Cancer and Bioactive Phytochemicals Inhibit the p53-MDM2 Pathway: A Computational Drug Development Approach. Molecules, 2023, 28(7): 2977.
DOI:10.3390/molecules28072977
8.
Jabin A, Uddin MF, Al Azad S, et al. Target-specificity of different amyrin subunits in impeding HCV influx mechanism inside the human cells considering the quantum tunnel profiles and molecular strings of the CD81 receptor: a combined in silico and in vivo study. In Silico Pharmacol, 2023, 11(1): 8.
DOI:10.1007/s40203-023-00144-6
9.
Arefin A, Gage MC. Metformin, Empagliflozin, and Their Combination Modulate Ex-Vivo Macrophage Inflammatory Gene Expression. Int J Mol Sci, 2023, 24(5): 4785.
DOI:10.3390/ijms24054785
10.
Morshed AKMH, Al Azad S, Mia MAR, et al. Oncoinformatic screening of the gene clusters involved in the HER2-positive breast cancer formation along with the in silico pharmacodynamic profiling of selective long-chain omega-3 fatty acids as the metastatic antagonists. Mol Divers, 2023, 27(6): 2651-2672.
DOI:10.1007/s11030-022-10573-8
11.
Dey D, Biswas P, Paul P, et al. Natural flavonoids effectively block the CD81 receptor of hepatocytes and inhibit HCV infection: a computational drug development approach. Mol Divers, 2023, 27(3): 1309-1322.
DOI:10.1007/s11030-022-10491-9
12.
Islam S, Farjana M, Uddin MR, et al. Molecular identification, characterization, and antagonistic activity profiling of Bacillus cereus LOCK 1002 along with the in-silico analysis of its presumptive bacteriocins. J Adv Vet Anim Res, 2022, 9(4): 663-675.
DOI:10.5455/javar.2022.i635
13.
Biswas P, Dey D, Biswas PK, et al. A Comprehensive Analysis and Anti-Cancer Activities of Quercetin in ROS-Mediated Cancer and Cancer Stem Cells. Int J Mol Sci, 2022, 23(19): 11746.
DOI:10.3390/ijms231911746
14.
Ferdausi N, Islam S, Rimti FH, et al. Point-specific interactions of isovitexin with the neighboring amino acid residues of the hACE2 receptor as a targeted therapeutic agent in suppressing the SARS-CoV-2 influx mechanism. J Adv Vet Anim Res, 2022, 9(2): 230-240.
DOI:10.5455/javar.2022.i588
15.
Dey D, Hasan MM, Biswas P, et al. Investigating the Anticancer Potential of Salvicine as a Modulator of Topoisomerase II and ROS Signaling Cascade. Front Oncol, 2022, 12: 899009.
DOI:10.3389/fonc.2022.899009
Table
2.
QSAR based bioactivity prediction for ligand validation
Compounds
Prediction of activity spectra for substances (Pa = 0.3 to 0.7)
Anti-infective
Anti-viral
Anti-microbial
Nobiletin
×
√
√
Reticuline
×
×
√
Kirenol
×
√
√
Chrysin
√
√
√
3-Caffeoylshikimic acid
√
√
√
Gigantamide A
×
√
×
Dasyclamide
×
√
√
QSAR: quantitative structure-activity relationship; Pa: prediction of activity score as per PASS server. Pa score of 0.3 to 0.7 signifies moderate activity. ×: Pa score less than 0.3; √: Pa score in the range 0.3 to 0.7
Table
5.
Solvent accessible surface area referring the area to energy ratio over the entire dynamic simulation process (100 nanoseconds) with polar and apolar regions precisely
Macromolecule
Ligands
WPR (Å)
GIC
TNR
Total area/energy (Ų/[kcal·mol])
Polar
Apolar
Unknown
α-DG
Nobiletin
1.4
No
244
4620.04
7575.65
0.00
α-DG
Reticuline
1.4
No
244
4620.04
7575.65
0.00
α-DG
Kirenol
1.4
No
244
4620.04
7575.65
0.00
α-DG
Chrysin
1.4
No
244
4620.04
7575.65
0.00
α-DG
3-caffeoylshikimic acid
1.4
No
244
4620.04
7575.65
0.00
α-DG
Gigantamide A
1.4
No
244
4620.04
7575.65
0.00
α-DG
Dasyclamide
1.4
No
244
4620.04
7575.65
0.00
WPR: water probe radius; GIC: gradient in calculation; TNR: total No. of residues.
 Authors and Reviewers
Authors and Reviewers


 DownLoad:
DownLoad: