Shen Tian, Han Bo'ang, Leng Yan, Yan Sen, Shi Junfeng, Yue Shen, Cheng Steven Y. Sonic Hedgehog stimulates migration of MCF-7 breast cancer cells through Rac1[J]. The Journal of Biomedical Research, 2019, 33(5): 297-307. DOI: 10.7555/JBR.32.20180100
Citation:
Shen Tian, Han Bo'ang, Leng Yan, Yan Sen, Shi Junfeng, Yue Shen, Cheng Steven Y. Sonic Hedgehog stimulates migration of MCF-7 breast cancer cells through Rac1[J]. The Journal of Biomedical Research, 2019, 33(5): 297-307. DOI: 10.7555/JBR.32.20180100
Shen Tian, Han Bo'ang, Leng Yan, Yan Sen, Shi Junfeng, Yue Shen, Cheng Steven Y. Sonic Hedgehog stimulates migration of MCF-7 breast cancer cells through Rac1[J]. The Journal of Biomedical Research, 2019, 33(5): 297-307. DOI: 10.7555/JBR.32.20180100
Citation:
Shen Tian, Han Bo'ang, Leng Yan, Yan Sen, Shi Junfeng, Yue Shen, Cheng Steven Y. Sonic Hedgehog stimulates migration of MCF-7 breast cancer cells through Rac1[J]. The Journal of Biomedical Research, 2019, 33(5): 297-307. DOI: 10.7555/JBR.32.20180100
Jiangsu Key Laboratory of Xenotransplanation, Department of Medical Genetics
2.
Department of Pathology and Pathophysiology
3.
Jiangsu Key Lab of Cancer Biomarkers, Prevention and Treatment, Collaborative Innovation Center for Cancer Personalized Medicine, Nanjing Medical University, Nanjing, Jiangsu 211166, China
4.
Department of Pathology, Jiangsu Province Hospital, Nanjing, Jiangsu 210029, China
5.
Department of Oncology, Nanjing First Hospital Affiliated to Nanjing Medical University, Nanjing, Jiangsu 210006, China
Steven Y Cheng and Shen Yue, Jiangsu Key Laboratory of Xenotransplanation, Department of Medical Genetics, Jiangsu Key Lab of Cancer Biomarkers, Prevention and Treatment, Collaborative Innovation Center for Cancer Personalized Medicine, Nanjing Medical University, Nanjing, Jiangsu 211166, China. Tel: + 86-25-86869463, E-mails: sycheng@njmu.edu.cn and yueshen@njmu.edu.cn
As one of the most common tumors in women, breast cancer has drawn considerable interest from investigators and clinicians in recent years. Despite early diagnosis and best therapeutic regimens available, the prognosis of malignant or metastatic breast cancer patients is still not optimistic. Hedgehog signaling, a classical pathway indispensable to embryonic development, participates in the growth of a variety of tumors. In the present study, the effect of Sonic Hedgehog (Shh) on breast cancer cells was investigated. We identified that Shh signal stimulated the migration of MCF-7 breast cancer cells. Smo and Gli1 were involved in Shh-stimulated migration of MCF-7 cells. Activating Smo and Gli1 induced cell migration, which was blocked by their specific antagonists. The effect of Shh signaling on MCF-7 cells was independent of Wnt5a, Dvl2 and Rab35, but directly dependent on Rac1. In conclusion, our study suggested that Shh promotes breast cancer cell migration via Rac1 independently of the non-canonical Wnt signaling pathway, which may represent a rational molecular target for combination medication in breast cancer.
Breast cancer is a most common cancer in women worldwide[1]. Although the prognosis of the primary tumors has improved significantly with new therapeutic techniques in recent years, the prognosis of metastatic breast cancer remains a major challenge in clinical work[2]. Tumor metastasis is a complex biological process, involving many genes and biomolecules[3]. Until now, the understanding of breast cancer metastasis is still incomplete. It is urgent to fully understand the biological basis of metastasis to improve the long-term survival of breast cancer patients[4].
The pseudopodium, which is mainly composed of lamellipodium and filopodium, participates in tumor metastasis. The formation of pseudopodium depends on microfilaments[5– 6], which are regulated by Rho family small GTPases. The Rac subfamily of Rho GTPases activates the front edge of cells. A representative member of Rac family is Rac1, which regulates the polymerization of the branched microfilaments, and thus plays a key role in the formation and function of lamellipodiums[7]. In our earlier study, Wnt5a, a nonclassical Wnt signal promotes the migration of MCF-7 breast cancer cells through Rac1[8]. However, migration involves many signaling pathways, so exploring the molecular mechanisms of migration is critical for precise selection of therapeutic targets during clinical treatment. An appropriate combination of multiple signaling pathway inhibitors can improve the efficacy of treatment and reduce the incidence of drug resistance.
It has been demonstrated that Sonic Hedgehog (Shh) signal is required for the growth and proliferation of a variety of tumors[9– 10]. Shh is an essential morphogenic and mitogenic factor in embryonic development and postnatal physiological processes. Shh signaling is initiated by the binding of Shh ligand to the 12-pass transmembrane receptor Patched-1 (Ptch1), which attenuates the inhibition of Ptch1 on Smoothened (Smo), a G-protein coupled receptor. Activated Smo turns on the downstream transcription program orchestrated by three transcription factors: Glioma-associated oncogene homolog 1, 2, and 3 (Gli1, Gli2 and Gli3)[11]. Mouse mammary cancer model studies have found the Shh-dependent mechanisms in mammary gland tumor formation and development[12]. Inhibition of Shh signaling reduces the survival of tumor cells and the abnormal stimulation of basal cells. However, it is not clear whether Shh contributes to metastasis of breast cancer.
In this study, we found that in MCF-7 breast cancer cells, Shh signal regulates the activation of Rac1, thereby affecting cell migration independent of Wnt5a, which called for further exploration of the potential crosstalk between two important signaling pathways of Wnt5a and Shh. It is also suggested that key molecules of the Shh signaling can be used as drug targets and combined with other drugs, to contribute to clinical breast cancer treatment.
Materials and methods
Cell culture
The human breast cancer cell line MCF-7 was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). MCF-7 cells were cultured in DMEM supplemented with 10% FBS and grown in a 5% CO2 atmosphere at 37 °C.
Tissue samples
Human normal and tumor tissues were obtained from the First Affiliated Hospital of Nanjing Medical University (Jiangsu Province Hospital). Written consent forms were obtained from all patients before tissue collection. All tissue samples were histologically confirmed with hemotoxylin-eosin staining. This study was approved by the Research Ethics Committee of Nanjing Medical University.
Actin cytoskeleton staining and immunofluorescence
Cells were fixed in 4% paraformaldehyde in PBS for 20 minutes, permeabilized in 0.1% Triton X-100 and blocked in PBS containing 1% BSA for 1 hour at room temperature. F-actin was stained with FITC-labeled phalloidin (5 μg/mL) (Sigma, St. Louis, MO, USA) for 60 minutes at 37 °C. After washing with PBS, the coverslips were mounted on glass slides with 4'-6- diamidino-2-phenylindole (DAPI) Fluoromount G (Southern Biotech, Birmingham, AL, US). The images were acquired with a fluorescence microscope (BX51, Nikon, Japan).
Wound-healing assay
MCF-7 cells were plated onto 96-well cell culture clusters (Costar, Cambridge, MA, USA) and grown to confluence, and then serum-starved for 24 hours. The transfected cells were cultured for 48 hours before serum starvation. The monolayer cells were scratched manually with a plastic pipette tip, and after two washes with PBS, the wounded cellular monolayer was allowed to heal for 24 hours in DMEM containing indicated compounds. Photographs of central wound edges were taken at time 0 and indicated time points using PowerShot G10 camera (Canon, Tokyo, Japan).
Cell migration assay
Cell migration was assessed in modified Boyden chamber assays (Costar), in which the two chambers were separated by a polycarbonate membrane (pore diameter, 8.0 mm). Boyden chamber wells were coated with human collagen type Ⅰ (10 mg/mL) for 1 hour at 37 °C. MCF-7 cells were grown to subconfluence in tissue culture plates and then suspended in serum-free medium supplemented with 5 mg/mL BSA. 5×104 cells were seeded into upper compartment. Medium containing indicated compounds was added to the upper and lower compartments of the Boyden chambers. The cells were allowed to migrate for 24 hours at 37 °C. Thereafter, the medium was discarded, stationary cells were removed with a cotton-tipped applicator and the membranes were cut out of the chamber and stained with 0.5% crystal violet. The response was evaluated in a light microscope by counting the number of cells that had migrated into the membrane.
Immunoblotting
Subconfluent cells were washed twice with PBS, and then lysed with ice-cold lysis buffer (50 mmol/L Tris, 150 mmol/L NaCl, 1% sodium deoxycholate, 1 mmol/L sodium orthovanadate, 1 mmol/L sodium fluoride, 1 mmol/L EDTA, 1 mmol/L PMSF, and 1% cocktail of protease inhibitors, pH7.4) containing the appropriate detergent for immunoblotting analysis of whole-cell lysates (1% SDS) or pulldown assays. The lysates were then clarified by centrifugation at 12 000 g for 20 minutes at 4 °C. The protein samples were separated by 8, 10, or 12% SDS-PAGE, transferred onto PVDF membranes, blocked with 5% milk-TBS-0.2% Tween, probed with primary antibodies, then horseradish peroxidase-conjugated secondary antibodies, and visualized with ECL reagent (Thermo Fisher Scientific, Waltham, MA, USA). The following primary antibodies were used: anti-GAPDH (Chemicon, Temecula, CA, USA), anti-HA (Thermo Fisher Scientific), anti-Rac1 (Proteintech, Chicago, IL, USA), anti-Shh (Abcam, Cambridge, MA, USA), and anti-Smo (Abcam) antibodies.
Active Rac1 pulldown assays
Endogenous active GTP-bound Rac1 levels were determined using PAK binding domain (PBD) pulldown assay. GST-PBD fusion protein was bound to MagneGST glutathione particles (Promega, Madison, WI, USA) as a bait to capture active Rac1. Cellular extracts were incubated with GST-PBD beads at 4 °C for 90 minutes. Thereafter, beads were captured magnetically, washed with ice-cold washing buffer (4.2 mmol/L Na2HPO4, 2 mmol/L KH2PO4, 280 mmol/ L NaCl, and 10 mmol/L KCl, pH7.2), and then resuspended in 2× Laemmli buffer. Levels of active and total Rac1 were detected by Western blotting.
Small interfering RNA (siRNA)
For gene knockdown, siRNA duplexes (Gene- Pharma, Shanghai, China) targeting Dvl2 (5′-GUGAGAGCUACCUAGUCAAT T-3 ′ and 5 ′-CGCUAAACAUGGAGAAGUATT-3 ′), Rab35 (5 ′- UUGAUUUCGUGAAGCCACC-3 ′), Rac1 (5 ′-GAGGAAGAGAAAAUGCCUGTT-3 ′ and 5 ′-GUUCUUAAUUUGCUUUUCCTT-3 ′), and negative control (N.C.) (5 ′-UUGUACUACACAAAAGUACUG-3 ′) were transfected into MCF-7 cells by using Lipofectamine 2000 Reagent as described in the previous section. Knockdown efficiency was evaluated by measuring protein levels in cell lysates after 48 hours of transfection.
Statistical analysis
Statistical analysis was performed with SPSS software package version 17 (SPSS Inc., Chicago, IL, USA). All data were performed as mean±SD. Twosided P values less than 0.05 were deemed statistically significant. The Chi-square test was applied to analyze the relationship between Shh expression status. Differences were considered as statistically significant data when P values were less than 0.05.
Results
Shh expression is higher in breast cancer tissues than normal tissues
It has been reported that the expression of Shh in breast cancers is higher than in normal mammary tissues[1]. TCGA database searching and UALCAN website analysis showed that Shh expression in tissues of luminal breast cancer, Her2 positive breast cancer and triple negative breast cancer was higher than that in normal breast tissues (Fig. 1A).
Figure
1.
Shh expression in breast cancer tissues is higher than normal tissues.
A: Expresssion of Shh in different subclasses of breast cancer (TCGA). Data were extracted from TCGA database and analyzed using the UALCAN website. B: H&E staining and immunohistochemical staining of Shh in normal breast tissues (left) and breast cancer tissues (right). Scale bar = 50 μm.
Further, we selected patient specimens with complete clinical records and pathological data for immunohistochemistry (IHC) from Department of Pathology of Jiangsu Province Hospital. The slices were divided into 6 groups according to relevant biomarkers to rule out potential inter-group differences (Table 1). IHC results showed that the expression of Shh was significantly increased in breast cancer compared to that in normal breast tissues (Fig. 1B), which is consistent with TCGA analysis.
Table
1.
Association between Shh and ER, PR, Her2 expression in breast tissues
Diagnosis
Case
Shha
P
Negative [n (%)]
Positive [n (%)]
Normal (n=20)
-
20
18 (90.0)
2 (10.0)
< 0.01
ER(–), PR(–), Her2(–)
19
0 (0.0)
19 (100.0)
ER(+), PR(+), Her2(–)
47
0 (0.0)
47 (100.0)
Tumor (n=100)
ER(–), PR(–), Her2(+)
16
0 (0.0)
16 (100.0)
ER(+), PR(+), Her2(+)
14
0 (0.0)
14 (100.0)
ER(+/–), PR(–/+), Her2(+/–)b
4
0 (0.0)
4 (100.0)
The differences between the two groups were evaluated by χ2 tests. aImmunoreactivity score of Shh: (–) as negative; (+)~(+++) as positive. bER(+/–), PR(–/+), Her2(+/–): ER(+), PR(–), Her2(+); ER(–), PR(+), Her2(+); ER(+), PR(–), Her2(–); ER(–), PR(+), Her2(–).
Shh stimulates MCF-7 breast cancer cell migration through Smo and Gli1 in vitro
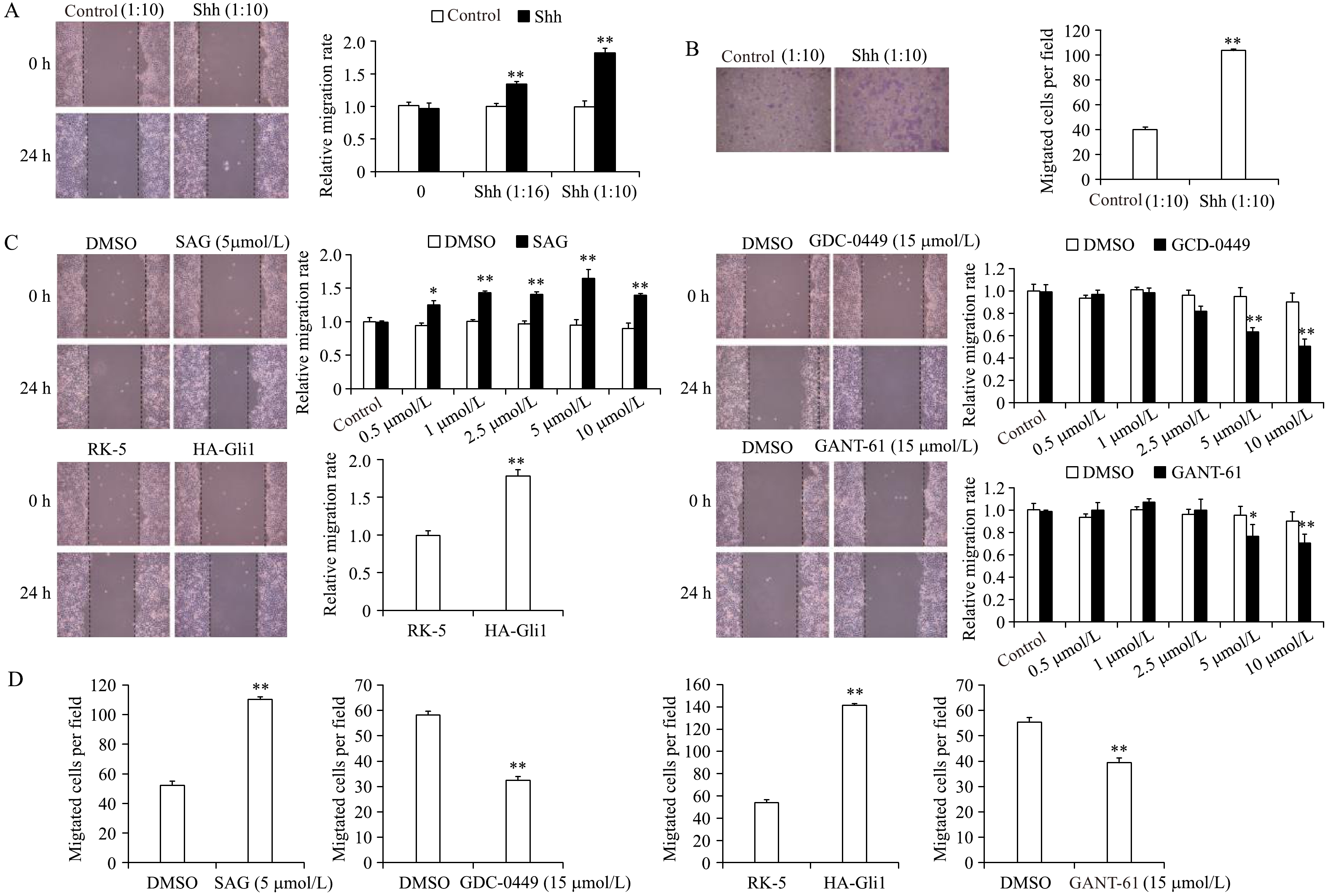
To understand the function of Shh in breast cancer, we treated MCF-7 breast cancer cells with Shh conditioned medium. The migration ability of MCF-7 cells was examined by wound healing assay and transwell migration assay. MCF-7 cells treated Shh ligand closed the wound faster than control cells (Fig. 2A). They showed significantly enhanced migration ability in Boyden chambers without collagen coat as well (Fig. 2B). These results indicated that Shh promotes the migration of MCF-7 cells.
Figure
2.
Shh stimulates MCF-7 breast cancer cell migration through Smo and Gli1 in vitro.
A and B: MCF-7 cells were stimulated by Shh ligand at the indicated doses for 24 hours. The cell motility rate was measured by wound-healing assay (A) or transwell migration assay (B). C, D: MCF-7 cells were stimulated by SAG, GDC-0449 or GANT-61 at the indicated doses for 24 hours, or transfected with HA-Gli1. The cell motility rate was measured by wound-healing assay (C) or transwell migration assay (D). *P<0.05, **P<0.01. Data are presented as mean±SD of 3 independent determinations.
Since Smo and Gli1 are both positive effectors of Shh signaling, we speculated if Smo and Gli1 could promote migration of MCF-7 cells. We incubated MCF-7 cells with different doses of Smo agonist (SAG), Smo inhibitor (vismodegib, GDC-0449) or Gli1 inhibitor (GANT-61), or transfected MCF-7 cells with HA-Gli1 plasmids, respectively. Those cells then underwent wound healing assay (Fig. 2C). The results showed that Smo activation and exogenous Gli1 promoted the MCF-7 migration, while the inhibition of Smo and Gli1 impeded it. We also performed transwell migration assay and got the consistent conclusion (Fig. 2D). These results identified that Shh signal promotes the migration of MCF-7 breast cancer cells through Smo and Gli1.
Shh induces Rac1 activation in MCF-7 breast cancer cells
Cytoskeleton drives shape changes during cell migration. Therefore, we incubated MCF-7 cells with control and Shh ligand, and then stained F-actin with FITC-phalliodin. Fluorescent staining indicated that Shh increased the quantity of stress fibers and lamellipodium of MCF-7 breast cancer cells (Fig. 3A). Rac1 was known to be involved in the information of lamellipodium, so we speculated that Rac1 activity might regulate the stimulation of cell migration by Shh in MCF-7 breast cancer cells.
Figure
3.
Shh induces Rac1 activation in MCF-7 breast cancer cells.
A: MCF-7 cells were treated with Shh ligand or control. Subsequently, cells were fixed and stained by FITC labeled phalloidin. B: Lysate was incubated with pGEX-2T (GST-Ctrl) protein which cannot bind to activated Rac1, or GST-PBD protein which can bind for GST-pulldown assay, to identify the specificity of GST-PBD. C: MCF-7 cells were treated with control or Shh ligand for different time periods, cell lysates were assayed for active Rac1 by pulldown assay using a GST-PBD as a bait. D–G: MCF-7 breast cancer cells were incubated for 24 hours with SAG (E), GDC-0449 (F) or GANT-61 (G) for indicated time, then were assayed for active Rac1 by pulldown assay using a GST-PBD as a bait. H: Active Rac1 pulldown assays of MCF-7 cells transfected with HA-Gli1. *P<0.05; **P<0.01. Data are presented as mean±SD of 3 independent determinations.
It was demonstrated in our earlier study that Rac1 regulated MCF-7 breast cancer cell migration as downstream of Wnt5a pathway[8]. According to the results above, a GST-pulldown assay (Fig. 3B) was proceeded to investigate Rac1 activation under Shh stimulation. Active Rac1 could be pulled down by GST fused Pak1 protein-binding domain (PBD) (Fig. 3B). We treated MCF-7 cells with Shh ligand for different time periods. Immunoblotting showed increased active Rac1 was pulled down from Shh treated cells. Activation of Rac1 peaked at 15 minutes of Shh treatment (Fig. 3C).
We further investigated the function of Smo and Gli1 on Rac1 activation in the same way. The results showed that SAG treatment and ectopic expression of HA-Gli1 significantly promoted Rac1 activation in MCF-7 cells, whereas GDC-0449 and GANT-61 had opposite effects (Fig. 3C–G). It indicated that Smo and Gli1 both positively regulated Rac1 activity.
Inhibition of Rac1 activation blocks the effect of Shh on the migration of MCF-7 breast cancer cells
To further verify the results above, we used siRNA to interfere Rac1 expression, or Rac GTPase inhibitor EHop-016 to block Rac1 activation, respectively. EHop-016 could significantly reduce the migration and the Rac1 activation of MCF-7 cells (Supplementary Fig. 1, available online), which was consistent with si-Rac1[8].
We found that Smo and Gli1 promoted cell migration and Rac1 activation of MCF-7 cells. However, SAG failed to promote the migration of Rac1-knockdown MCF-7 in wound healing assay and transwell migration assay. And HA-Gli1failed as well (Fig. 4A and 4B). EHop-016 completely blocked the effect of SAG and exogenous Gli1 on Rac1 activation in MCF-7 cells (Fig. 4C and 4D).
Figure
4.
Inhibition of Rac1 activation blocks the effect of Shh on the migration of MCF-7 breast cancer cells.
A and B: MCF-7 cells were incubated with SAG or transfected with HA-Gli1 after Rac1 siRNA transfection; cell migration rate was detected by wound-healing (A) and transwell assays (B). C and D: MCF-7 cells were incubated with SAG (C) or transfected with HA-Gli1 (D) after EHop-016 treatment, and cellular lysates were assayed for active Rac1 by pulldown assay using a GST-PBD as a bait. **P<0.01, compared to the untreated group without transfection; ##P<0.01, compared to EHop-016 untreated group with SAG or HA-Gli1. Data are presented as mean±SD of 3 independent determinations.
So far, we confirmed that Shh could promote the migration of MCF-7 breast cancer cells by up-regulating Rac1 activation.
Shh regulates MCF-7 cell migration and Rac1 activation independently on Wnt5a
Our published study showed that Wnt5a controled the activation of Rac1 on MCF-7 cell migration through Dvl2 and Rab35[8]. Since Shh could also regulate Rac1 activation, we were curious about the crosstalk between Shh and Wnt5a pathway in cell migration. In search of an answer, we knocked down Dvl2 and Rab35 respectively in MCF-7 cells by siRNA, then treated the cells with SAG or exogenously expressed Gli1. The results showed that the siRNA reduced the migration rate (Fig. 5A and 5B, 5E and 5F) and Rac1 activation (Fig. 5C and 5D, 5G and 5H), which were both reversed by SAG and HA-Gli1. These results suggested that Shh ddi not depend on the Wnt5a-Dvl2-Rab35 axis to regulate Rac1 activation.
Figure
5.
Shh signaling regulates MCF-7 breast cancer cell migration and Rac1 activation independent of Wnt5a.
A and B: MCF-7 breast cancer cells were incubated with SAG or GDC-0449 after Dvl2 or Rab35 siRNA transfection, cell migration rate was detected by wound-healing and transwell assays (B). C and D: cellular lysates were assayed for active Rac1 by pulldown assay using a GST-PBD as a bait. E and F: Overexpressing Gli1 in MCF-7 breast cancer cells after Dvl2 or Rab35 siRNA transfection, cell migration rate was detected by wound-healing and transwell assays. G and H: Cellular lysates were assayed for active Rac1 by pulldown assay using a GST-PBD as a bait. **P<0.01. Data are presented as mean±SD of 3 independent determinations.
In summary, we present the evidence that both Wnt5a and Shh promote breast cancer cell migration via Rac1 (Fig. 6). These findings highlight the presence of a potential molecular mechanism of resistance, which may represent a rational molecular target for combination medication in breast cancer.
Figure
6.
Model for the role of Wnt5a and Shh in promoting MCF-7 breast cancer cell migration by activating Rac1.
The world known cases of breast cancer has reached about 1.4 million[13], and the incidence of invasive breast cancer in women has been up to 1/8, in the forefront of the list of cancer-related death. Although early diagnosis and adjuvant treatment of breast cancer have been more and more emphasized in recent years, the prognosis malignant or metastatic breast cancer is still not optimistic[14]. Researchers and clinicians carried out extensive research to explore the carcinogenic mechanisms of breast cancer, expecting to find more effective therapeutic targets and biomarkers to predict prognosis more accurately. Our previous study proved that non-canonical Wnt5a signaling plays a role in the migration of MCF-7 breast cancer cells. The Wnt family, which is indispensable in embryonic development like Shh, has been demonstrated to participate in tumorigenesis nowadays. The function of Wnt5a depends on the availability of key receptors and intercellular interactors in tumor cells[15]. It stimulates the cell migration through a Dvl2-Rab35-Rac1 axis in MCF-7 breast cancer cells[8].
In recent years, the role of Hedgehog (Hh) in tumorigenesis gradually came to the surface[16– 18]. Recent studies have confirmed that the Hh signaling pathway plays a significant role in breast cancer progression, although the mechanism is not very clear[19]. Hh signaling conservatively participates in embryonic development process of invertebrates and vertebrates[20]. Most adults have no or very low Hh signaling activity. It was activated only when necessary, such as repairing tissue injuries[21– 23]. Although Hh signaling is integral to embryonic development, the excessive activation of it might lead to diseases, including cancer[9,24– 25]. Mutation and overactivation of Hh signaling pathway are associated with basal cell carcinoma (BCC), medulloblastoma (MB), breast cancer, pancreatic cancer, prostate cancer and lung cancer, etc[10].
Mammary gland development can be divided into three stages: embryo, non-pregnancy and pregnancy/ lactation[26]. The Hh signaling was detected in mouse breast tissues in all the stages. However, the experimental results show that Hh signaling pathway is not decisive in mice mammary gland development before or after birth. As a matter of fact, inhibition of Hh signaling is found in the normal mammary gland development. Lack of Gli1 and Gli2 gene expression does not lead to obvious mammary duct defects in embryos, while adult mice with sustained activation of Gli1 or lack of GLI3 function have mammary duct defects[27]. The overexpression of Shh in embryos of transgenic mouse, sustained activation of SMO or lack of PTCH1 may lead to abnormal breast terminal end buds (TEBs) morphology, which can be linked to human mammary gland hyperplasia[28– 29]. Correspondingly, overexpression of Gli1 in the mouse mammary epithelium leads to defects in the mammary duct network, making it unable to lactate, prone to hyperplasia, and thus increase the cancer risk[30]. In brief, aberrant activation of Hh signaling leads to disordered breast development and promotes breast cancer.
The potential role of Hh signaling in breast cancer is not clear yet. Kubo[31] first reported that SHH, PTCH1, and Gli1 were detected in the invasive breast cancer tissue, but not in the normal breast tissue. The expression of Hh ligand in epithelial cells occurs in early breast cancer and is closely related to the basal type phenotype, which is prone to metastasis, poor prognosis and high mortality. In addition, compared with normal mice, the over-expression of Hh ligand in basal subtype breast cancer (BLBC) mouse models leads to faster tumor growth and higher invasiveness. A recent study by Ramaswamy found that the nonclassical Hh signaling pathways contributed to continuous growth of tamoxifen-resistant breast cancer cells[32].
Despite advances in radiation therapy, chemotherapy and hormone therapy in recent years, 20% to 30% of early breast cancer patients still suffer in-situ recurrences or distant metastases[33]. In particular, BLBC subtype patients have a higher risk of distant metastasis and a shorter interval from metastasis to death, with only 50% of 10-year survival rate[34]. Clinic trials showed that targeted therapy is the most effective way, for example: estrogen receptor (ER) inhibitors (tamoxifen) and HER2 inhibitors (Trastuzumab) for ER and HER2 positive cases. However, ER and HER2 expressions are usually negative in typical BLBC patients, so they are usually not sensitive to standardized chemotherapy. Therefore, due to the lack of specific clinical therapeutic target, BLBC usually has poor prognosis. To reduce the mortality of this invasive breast cancer, new therapeutic targets are imperative. Experiments show that, as in esophageal and pancreatic cancer, the Hh signaling pathway, as an effective pharmacological target of BLBC, can consolidate the curative effect of chemotherapy and/or radiotherapy[35– 36]. Accurate selection of suitable patients and the drug combination are key to molecular target therapy for breast cancer. If Hh is to be used as a therapeutic target, it is urgent to clarify the cellular and molecular characteristics of Hh pathway in breast cancer.
The Hh signaling pathway can be inactivated at different cascaded levels, from blocking Hh ligands secretion to inhibiting Gli function with antibodies[37]. The most widely used inhibitors of Hh signaling pathway are Smo antagonists. Cyclopamine and jervine are two firstly-found Smo inhibitors. High throughput sequencing in vitro also identified more effective Smo inhibitors, such as vismodegib[38–40] and sonidegib, which were confirmed in advanced basal cell carcinoma. However, some cases of resistance appeared in vismodegib and sonidegib treatment. A major manifestation of drug resistance is the reactivation of the Shh pathway, which is likely to be associated with multiple pathways that block the carcinogenic signaling pathway in many tumors[41–43].
To sum up, data have confirmed that the Shh activity is inhibited during development and functional operation of the mammary gland; while in invasive breast cancer, such as BLBC, it is activated and participates in tumor growth. Here we found that Shh promoted breast cancer cell migration via Rac1, suggesting it is an available target of breast cancer therapy. Therefore, identifying the invasion and metastasis mechanisms mediated by Hh signaling pathway in breast cancer cells, and the crosstalk between other pathways, such as Wnt pathways, will help to improve the prognosis of breast cancer.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (No. 81672748 and 81871936 to S.Y.C.; No. 81572720 to S.Y.).
Jemal A, Bray F, Center MM, et al. Global cancer statistics[J]. CA Cancer J Clin, 2011, 61(2): 69–90. doi: 10.3322/caac.v61:2
[2]
Lorusso G, Rüegg C. New insights into the mechanisms of organ-specific breast cancer metastasis[J]. Semin Cancer Biol, 2012, 22(3): 226–233. doi: 10.1016/j.semcancer.2012.03.007
[3]
Spano D, Heck C, De Antonellis P, et al. Molecular networks that regulate cancer metastasis[J]. Semin Cancer Biol, 2012, 22(3): 234–249. doi: 10.1016/j.semcancer.2012.03.006
[4]
Ding L, Ellis MJ, Li S, et al. Genome remodelling in a basallike breast cancer metastasis and xenograft[J]. Nature, 2010, 464(7291): 999–1005. doi: 10.1038/nature08989
[5]
Brooks SA, Lomax-Browne HJ, Carter TM, et al. Molecular interactions in cancer cell metastasis[J]. Acta Histochem, 2010, 112(1): 3–25. doi: 10.1016/j.acthis.2008.11.022
[6]
Christofori G, Semb H. The role of the cell-adhesion molecule E-cadherin as a tumour-suppressor gene[J]. Trends Biochem Sci, 1999, 24(2): 73–76. doi: 10.1016/S0968-0004(98)01343-7
[7]
Zegers MM, Friedl P. Rho GTPases in collective cell migration[J]. Small GTPases, 2014, 5: e28997.
[8]
Zhu Y, Shen T, Liu J, et al. Rab35 is required for Wnt5a/Dvl2- induced Rac1 activation and cell migration in MCF-7 breast cancer cells[J]. Cell Signal, 2013, 25(5): 1075–1085. doi: 10.1016/j.cellsig.2013.01.015
[9]
Wu F, Zhang Y, Sun B, et al. Hedgehog signaling: from basic biology to cancer therapy[J]. Cell Chem Biol, 2017, 24(3): 252–280. doi: 10.1016/j.chembiol.2017.02.010
[10]
Amakye D, Jagani Z, Dorsch M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer[J]. Nat Med, 2013, 19(11): 1410–1422. doi: 10.1038/nm.3389
[11]
Pak E, Segal RA. Hedgehog signal transduction: key players, oncogenic drivers, and cancer therapy[J]. Dev Cell, 2016, 38(4): 333–344. doi: 10.1016/j.devcel.2016.07.026
[12]
Mun Hui, Aurélie Cazet, Radhika Nair1, et al. The Hedgehog signalling pathway in breast development, carcinogenesis and cancer therapy[J]. Breast Cancer Res, 2013, 15: 203. doi: 10.1186/bcr3401
[13]
Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries[J]. CA Cancer J Clin, 2018, 68(6): 1–31.
[14]
Ferlay J, Shin HR, Bray F, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008[J]. Int J Cancer, 2010, 127(12): 2893–2917. doi: 10.1002/ijc.v127:12
[15]
Zeng R, Huang J, Zhong MZ, et al. Multiple roles of WNT5A in breast cancer[J]. Med Sci Monit, 2016, 22: 5058–5067. doi: 10.12659/MSM.902022
[16]
Karhadkar SS, Bova GS, Abdallah N, et al. Hedgehog signalling in prostate regeneration, neoplasia and metastasis[J]. Nature, 2004, 431(7009): 707–712. doi: 10.1038/nature02962
[17]
Michaud EJ, Yoder BK. The primary cilium in cell signaling and cancer[J]. Cancer Res, 2006, 66(13): 6463–6467. doi: 10.1158/0008-5472.CAN-06-0462
[18]
Watkins DN, Berman DM, Burkholder SG, et al. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer[J]. Nature, 2003, 422(6929): 313–317. doi: 10.1038/nature01493
[19]
Benvenuto M, Masuelli L, De Smaele E, et al. In vitro and in vivo inhibition of breast cancer cell growth by targeting the Hedgehog/GLI pathway with SMO (GDC-0449) or GLI (GANT-61) inhibitors[J]. Oncotarget, 2016, 7(8): 9250–9270.
[20]
Varjosalo M, Taipale J. Hedgehog: functions and mechanisms[J]. Genes Dev, 2008, 22(18): 2454–2472. doi: 10.1101/gad.1693608
[21]
Beachy PA, Karhadkar SS, Berman DM. Tissue repair and stem cell renewal in carcinogenesis[J]. Nature, 2004, 432(7015): 324–331. doi: 10.1038/nature03100
[22]
Palma V, Ruiz i Altaba A. Hedgehog-GLI signaling regulates the behavior of cells with stem cell properties in the developing neocortex[J]. Development, 2004, 131(2): 337–345.
[23]
Kaustio M, Haapaniemi E, Göös H, et al. Damaging heterozygous mutations in NFKB1 lead to diverse immunologic phenotypes[J]. J Allergy Clin Immunol, 2017, 140(3): 782–796. doi: 10.1016/j.jaci.2016.10.054
[24]
Hahn H, Wicking C, Zaphiropoulous PG, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome[J]. Cell, 1996, 85(6): 841–851. doi: 10.1016/S0092-8674(00)81268-4
[25]
Romer JT, Kimura H, Magdaleno S, et al. Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(+/-)p53(-/-) mice[J]. Cancer Cell, 2004, 6(3): 229–240. doi: 10.1016/j.ccr.2004.08.019
[26]
Watson CJ, Khaled WT. Mammary development in the embryo and adult: a journey of morphogenesis and commitment[J]. Development, 2008, 135(6): 995–1003. doi: 10.1242/dev.005439
[27]
Hatsell SJ, Cowin P. Gli3-mediated repression of Hedgehog targets is required for normal mammary development[J]. Development, 2006, 133(18): 3661–3670. doi: 10.1242/dev.02542
[28]
Moraes RC, Zhang X, Harrington N, et al. Constitutive activation of smoothened (SMO) in mammary glands of transgenic mice leads to increased proliferation, altered differentiation and ductal dysplasia[J]. Development, 2007, 134(6): 1231–1242. doi: 10.1242/dev.02797
[29]
Lewis MT, Ross S, Strickland PA, et al. Defects in mouse mammary gland development caused by conditional haploinsufficiency of Patched-1[J]. Development, 1999, 126(22): 5181–5193.
[30]
Fiaschi M, Rozell B, Bergström A, et al. Targeted expression of GLI1 in the mammary gland disrupts pregnancy-induced maturation and causes lactation failure[J]. J Biol Chem, 2007, 282(49): 36090–36101. doi: 10.1074/jbc.M704280200
[31]
Kubo M, Nakamura M, Tasaki A, et al. Hedgehog signaling pathway is a new therapeutic target for patients with breast cancer[J]. Cancer Res, 2004, 64(17): 6071–6074. doi: 10.1158/0008-5472.CAN-04-0416
[32]
Xuan Y, Lin Z. Expression of Indian Hedgehog signaling molecules in breast cancer[J]. J Cancer Res Clin Oncol, 2009, 135(2): 235–240. doi: 10.1007/s00432-008-0451-x
[33]
Heller E, Hurchla MA, Xiang J, et al. Hedgehog signaling inhibition blocks growth of resistant tumors through effects on tumor microenvironment[J]. Cancer Res, 2012, 72(4): 897–907. doi: 10.1158/0008-5472.CAN-11-2681
[34]
Das S, Tucker JA, Khullar S, et al. Hedgehog signaling in tumor cells facilitates osteoblast-enhanced osteolytic metastases[J]. PLoS One, 2012, 7(3): e34374. doi: 10.1371/journal.pone.0034374
[35]
Early Breast Cancer Trialists' Collaborative Group (EBCTCG). Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials[J]. Lancet, 2005, 365(9472): 1687–1717. doi: 10.1016/S0140-6736(05)66544-0
[36]
Kennecke H, Yerushalmi R, Woods R, et al. Metastatic behavior of breast cancer subtypes[J]. J Clin Oncol, 2010, 28(20): 3271–3277. doi: 10.1200/JCO.2009.25.9820
[37]
El-Zaatari M, Kao JY, Tessier A, et al. Gli1 deletion prevents Helicobacter-induced gastric metaplasia and expansion of myeloid cell subsets[J]. PLoS One, 2013, 8(3): e58935. doi: 10.1371/journal.pone.0058935
[38]
Cochrane CR, Szczepny A, Watkins DN, et al. Hedgehog signaling in the maintenance of cancer stem cells[J]. Cancers (Basel), 2015, 7(3): 1554–1585. doi: 10.3390/cancers7030851
[39]
Balbous A, Renoux B, Cortes U, et al. Selective release of a cyclopamine glucuronide prodrug toward stem-like cancer cell inhibition in glioblastoma[J]. Mol Cancer Ther, 2014, 13(9): 2159–2169. doi: 10.1158/1535-7163.MCT-13-1038
[40]
Liu S, Dontu G, Mantle ID, et al. Hedgehog signaling and Bmi- 1 regulate self-renewal of normal and malignant human mammary stem cells[J]. Cancer Res, 2006, 66(12): 6063–6071. doi: 10.1158/0008-5472.CAN-06-0054
[41]
Moraes RC, Chang H, Harrington N, et al. Ptch1 is required locally for mammary gland morphogenesis and systemically for ductal elongation[J]. Development, 2009, 136(9): 1423–1432. doi: 10.1242/dev.023994
[42]
McDermott KM, Liu BY, Tlsty TD, et al. Primary cilia regulate branching morphogenesis during mammary gland development[J]. Curr Biol, 2010, 20(8): 731–737. doi: 10.1016/j.cub.2010.02.048
[43]
Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2[J]. N Engl J Med, 2001, 344(11): 783–792. doi: 10.1056/NEJM200103153441101
Table
1.
Association between Shh and ER, PR, Her2 expression in breast tissues
Diagnosis
Case
Shha
P
Negative [n (%)]
Positive [n (%)]
Normal (n=20)
-
20
18 (90.0)
2 (10.0)
< 0.01
ER(–), PR(–), Her2(–)
19
0 (0.0)
19 (100.0)
ER(+), PR(+), Her2(–)
47
0 (0.0)
47 (100.0)
Tumor (n=100)
ER(–), PR(–), Her2(+)
16
0 (0.0)
16 (100.0)
ER(+), PR(+), Her2(+)
14
0 (0.0)
14 (100.0)
ER(+/–), PR(–/+), Her2(+/–)b
4
0 (0.0)
4 (100.0)
The differences between the two groups were evaluated by χ2 tests. aImmunoreactivity score of Shh: (–) as negative; (+)~(+++) as positive. bER(+/–), PR(–/+), Her2(+/–): ER(+), PR(–), Her2(+); ER(–), PR(+), Her2(+); ER(+), PR(–), Her2(–); ER(–), PR(+), Her2(–).
 Authors and Reviewers
Authors and Reviewers


 DownLoad:
DownLoad: